


Podiel pacientov s imunodeficienciou stúpa najmä v dôsledku nárastu chronických chorôb vyžadujúcich imunosupresívnu alebo biologickú liečbu. V menšej miere zaznamenávame tiež nárast primárnych (intrinzických) porúch imunity, ktorý súvisí s ich zlepšujúcou sa diagnostikou a liečbou. S dĺžkou trvania imunodeficiencie sa pritom zvyšuje nielen riziko infekčných komplikácií, ale aj riziko malígnej transformácie buniek a tkanív. Podrobnejšie skúsenosti o tom, ktoré poruchy imunity sú v tomto ohľade najrizikovejšie priniesol práve výskum primárnych imunodeficiencií v posledných rokoch.
Imunodeficiencie predstavujú veľkú skupinu porúch, ktoré sú spôsobené poruchou funkcie jednej alebo viacerých zložiek imunitného systému. Rozdeľujeme ich na primárne a sekundárne. Príčina primárnych imunodeficiencií spočíva v genetickom defekte buniek alebo proteínov kľúčových pre správnu funkciu imunity. Vo väčšine prípadov ide o monogénové poruchy, ktoré sa môžu manifestovať kedykoľvek v priebehu života. Menšiu časť tvoria poruchy, u ktorých dosiaľ nebol identifikovaný genetický defekt a predpokladá sa polygénový charakter dedičnosti. Primárne imunodeficiencie sa obvykle považujú za zriedkavé choroby a jednotlivé choroby aj nimi sú. Týchto viac než 230 dosiaľ identifikovaných porúch predstavuje významnú skupinu ochorení, ktoré bez liečby ovplyvňujú životy relatívne veľkého počtu pacientov. Sekundárne imunodeficiencie, ktoré sú približne desaťnásobne častejšie, sú podmienené vonkajšími faktormi, medzi ktoré patria vírusové infekcie (najvýznamnejšia HIV infekcia), malígne lymfoproliferatívne a myeloproliferatívne ochorenia, ťažký stres ako dôsledok operácie alebo traumy, enterálne a renálne straty proteínov, ťažká malnutrícia a mnohé ďalšie okolnosti vrátane farmák (najmä imunosupresív).
Imunita a klasifikácia jej primárnych porúch
Základnou funkciou imunitného systému je chrániť organizmus pred infekciou vyvolanou mikroorganizmami rôzneho druhu, ako sú baktérie, vírusy, plesne alebo protozoá. Následkom poruchy sú imunodeficitní pacienti náchylní na infekčné komplikácie aj málo virulentnými alebo nepatogénnymi organizmami. Chybná regulácia imunitného systému – ako dôsledok defektu niektorej zložky – môže spôsobiť, že sa imunitná odpoveď obráti proti vlastným tkanivám. Výsledkom je chronický zápal a autoimunita. Ďalším dôsledkom chybnej regulácie je zlyhanie imunologického dohľadu a malígna transformácia buniek a tkanív. Podobný dôsledok má nefunkčná protivírusová ochrana v prípade infekcií onkogénnymi vírusmi. Imunodeficiencia môže byť rovnako dôsledkom ako príčinou malígneho ochorenia. Ďalej sa podrobnejšie budeme zaoberať primárnymi imunodeficienciami (PID), ktoré sú spojené so zvýšeným rizikom malígneho postihnutia.
Komplikácie imunodeficiencií
Infekčné komplikácie sú najčastejším klinickým prejavom imunodeficiencie. Príznaky nie sú obvykle špecifické. Môže ísť o opakujúce sa alebo chronicky prebiehajúce respiračné infekcie, gastrointestinálne infekcie, chronické zápalové postihnutie kĺbov, pečene, kože alebo iných orgánov. Často ostávajú nerozpoznané alebo rozpoznané neskoro, keď sú už prítomné orgánové zmeny. Akútne infekcie nie sú vždy iniciálnymi príznakmi. Medzi druhú najčastejšiu príčinu úmrtia pacientov s PID patria malígne ochorenia. Celkové riziko malígnych ochorení spojených s PID sa odhaduje v rozmedzí 4 – 25 %, pre niektoré podskupiny PID môže byť ale oveľa vyššie. Zlepšujúca sa diagnostika imunodeficiencií a na ňu nadväzujúca liečba, najmä efektívna substitúcia imunoglobulínov IgG, vedie k významnému poklesu infekčných komplikácií. Vďaka tomu sa pacienti dožívajú podstatne vyššieho veku a riziko malígnych ochorení stúpa. Substitúcia imunoglobulínmi nedokáže nahradiť všetky izotypy imunoglobulínov a nedokáže ani ovplyvniť poruchy celulárnej imunity. Tie majú zásadný vplyv na priebeh najmä vírusových infekcií, ktoré pokračujú asymptomaticky. Porucha imunitného dohľadu zvyšuje vnímavosť na nové infekcie, alebo vedie k reaktivácii latentnej alebo chronickej infekcie. Postihnuté sú najmä bunky s vysokým antigénnym potenciálom, preto najčastejšou formou malígnych ochorení u imunodeficientných pacientov sú lymfómy, t. j. malígne choroby imunitného systému.
Odhaduje sa, že až pätina lymfómov u imunodeficientných pacientov je indukovaná vírusom Ebstein-Barrovej (EBV). Infekcia týmto humánnym herpes vírusom (HHV-4) patrí medzi najčastejšie vírusové infekcie u detí aj dospelých. Viac než 90 percent dospelých má prítomné pamäťové protilátky proti tomuto vírusu, čo dokazuje kontakt imunitného systému s vírusom. Na rozdiel od zdravej populácie majú imunodeficientní pacienti zvýšenú náchylnosť na komplikácie. Vírus vstupuje do organizmu najčastejšie orálnou kvapôčkovou nákazou. Hoci môže infikovať epitelové bunky a T lymfocyty, má špecifickú afinitu k B lymfocytom, ktoré slúžia ako jeho rezervoár. EBV pôsobí silne proliferačne a stimuluje tvorbu špecifických aj nešpecifických protilátok. Krátko po začiatku infekcie sa aktivuje cytotoxická imunitná odpoveď. Defekt v celulárnej T lymfocytovej imunite umožňuje nekontrolovanú proliferáciu B lymfocytov. U pacientov s primárnou imunodeficienciou, postihujúcou špecifickú celulárnu imunitu, spôsobuje rôzne lymfoproliferatívne poruchy B lymfocytového radu vrátane Burkittovho lymfómu, klasického Hodgkinovho lymfómu alebo potransplantačnú lymfoproliferatívnu chorobu. Lymfoproliferatívna choroba viazaná na chromozóm X (XLP) je porucha signálneho proteínu SH2D1A, ktorá spôsobuje poruchu cytotoxickej funkcie lymfocytov. Po primoinfekcii EBV sa u dovtedy zdravých chlapcov vyvíja obraz ťažkej infekčnej mononukleózy, často s fatálnym koncom. U tých, ktorí prežijú, sa vyvíja hypogamaglobulinémia alebo malígny lymfóm. Za takmer polovicou prípadov Hodgkinovej choroby stojí infekcia EBV.
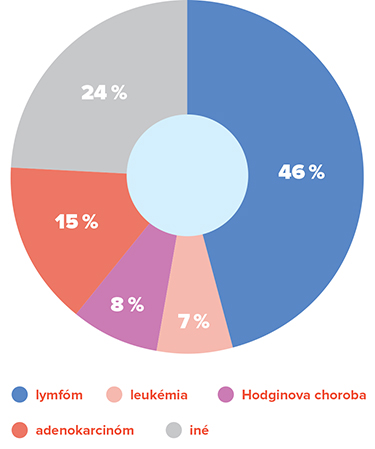
Najčastejšie malígne choroby u imunokompromitovaných pacientov
Takmer polovicu malígnych chorôb u pacientov s primárnymi imunodeficienciami tvorí non-Hodgkinov lymfóm (NHL), ktorý je najrozšírenejším druhom rakoviny lymfatického systému vôbec (3). Druhou najčastejšou malígnou chorobou, postihujúcou približne 10 % pacientov s imunodeficienciou, je Hodgkinov lymfóm (4). Doteraz najväčšou populačnou štúdiou, ktorá skúmala vzťah medzi PID a malígnymi chorobami, bola práca austrálskych autorov (5). Autori prepojili údaje od 1 132 pacientov z austrálskeho registra PID s národným austrálskym registrom zomrelých a registrom malígnych chorôb. Jednotliví pacienti boli sledovaní v priemere 16 rokov. Za toto obdobie bolo relatívne riziko vzniku rakoviny 1,6-násobne vyššie, súhrnne u všetkých typov PID. Najčastejším malígnym ochorením bol NHL, následne leukémie a rakovina žalúdka. Štúdia potvrdila zvýšené riziko rakoviny v kategórii protilátkových imunodeficiencií (tvorili 78 % súboru) a v kategórii iných dobre definovaných imunodeficitných syndrómov. Na rozdiel od T bunkových imunodeficiencií sa prevažne protilátkové imunodeficiencie spájali s užším spektrom solídnych tumorov.
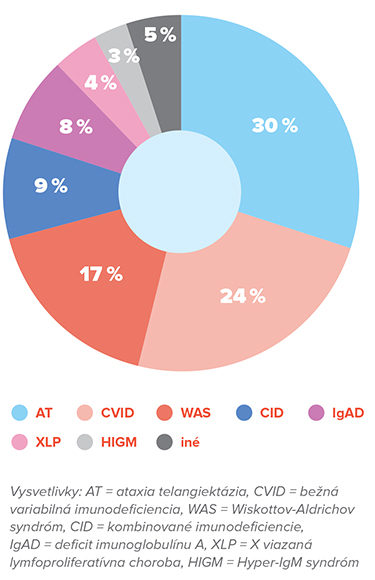
Humorálne imunodeficiencie spojené s malígnym ochorením
Najčastejšou klinicky významnou poruchou špecifickej humorálnej imunity u detí aj dospelých je bežná variabilná imunodeficiencia (CVID). V súčasnosti považujeme toto ochorenie za heterogénnu skupinu s rôznym klinickým priebehom a prognózou. Časť postihnutých má dominantne endogénnu poruchu B lymfocytov, u viac ako tretiny sa pozoruje porucha aj na úrovni T lymfocytov. Približne 10 % má výlučne infekčné komplikácie. U ostatných sa objavujú autoimunitné komplikácie rôzneho charakteru, chronické granulomatózne zápalové procesy, splenomegália a lymfoproliferácia. Len u približne 10 % sa podarilo identifikovať molekulový defekt (ICOS, CD19, BAFF-R, TNFRSF13B alebo CD81) a tieto poruchy novšia klasifikácia vyčleňuje z kategórie CVID. Častejší výskyt CVID v príbuzenstve potvrdzuje dedičný charakter, predpokladá sa polygénový typ.
Klinicky sa porucha prejavuje chronickými infekciami horných a dolných dýchacích ciest, prevažne bakteriálnej etiológie. V laboratórnom obraze majú pacienti znížené hodnoty IgG (pod 2SD pre daný vek) a súčasne znížené alebo chýbajúce IgA a/alebo IgM. Incidencia je najvyššia v mladom dospelom veku, u detí nie skôr ako po 4. roku života. Z malígnych komplikácií sú najčastejšie NHL a karcinóm žalúdka. Vysoká incidencia rakoviny žalúdka sa dáva do spojitosti s kolonizáciou Helicobacter pylori, ľudským herpes vírusom 8 a poškodením p53, celulárnym tumor supresorovým antigénom (transkripčný faktor zabraňujúci vzniku nádorov). Výskyt rakoviny u CVID pacientov diagnostikovaných v dospelosti je viac než 3-násobne vyšší v porovnaní s detskými pacientmi. Podľa jednej štúdie sa malígne ochorenie vyskytlo u 11 % pacientov v priebehu 13 rokov sledovania (6). Podľa tejto štúdie tvorili väčšinu ženy v 5. – 6. dekáde života. Riziko NHL bolo u pacientov s CVID v porovnaní s bežnou populáciou 30 až 400-násobné. Podľa vyššie spomínanej austrálskej štúdie mali pacienti s CVID 7-násobne zvýšené riziko rakoviny žalúdka, riziko NHL bolo 12-násobne vyššie a riziko neoplázie týmusu až 146-násobne vyššie. Spojenie tymómu a imunodeficiencie sa označuje ako Goodov syndróm. Ide o samostatnú klinickú jednotku, ktorá sa vyskytuje v 4. až 5. dekáde života. Dôsledkom chronickej zápalovej alebo autoimunitnej stimulácie sa môže u pacientov s CVID vyvinúť MALT lymfóm (lymfóm asociovaný so slizničným lymfoidným tkanivom). Ide o NHL nízkeho stupňa vychádzajúci z B lymfocytov. Prognóza malígnych komplikácií u CVID pacientov sa vo všeobecnosti považuje za porovnateľnú s imunokompetentnými pacientmi.
Najčastejšou primárnou imunodeficienciou vo všeobecnosti je deficit IgA (IgAD). Prevalencia sa odhaduje na 1 prípad na 500 jedincov. Prevažná väčšina pacientov je asymptomatická, a preto mnohí o poruche ani nevedia. Pravdepodobne u tretiny postihnutých sú prítomné symptómy opakujúcich sa respiračných a gastrointestinálnych infekcií, alergické choroby, autoimunitné choroby, lymfoproliferatívne poruchy a orgánové malignity prevažne gastrointestinálneho traktu. IgAD a CVID majú viacero príbuzných znakov. U časti IgAD pacientov sa neskôr vyvíja CVID, a preto sa tieto poruchy považujú za spektrum chorôb rovnakého genetického základu. Riziko malignity u IgAD je nižšie v porovnaní s CVID. Keďže mnohí onkologickí pacienti majú pokles IgA, špekuluje sa, či nemôže ísť aj o primárnu poruchu IgAD a nie sekundárne indukovanú hypogamaglobulinémiu.
Celulárne imunodeficiencie spojené s malígnym ochorením
Táto skupina porúch je veľmi rôznorodá, čo sa týka mechanizmu imunodeficiencie a klinického obrazu. Wiskottov-Aldrichov syndróm je na X chromozóm viazaná imunodeficiencia, ktorej hlavnými diagnostickými znakmi sú trombocytopénia, ekzém a bakteriálne infekcie. Krvácavé prejavy v podobe petechií a hematómov v dôsledku trombocytopénie sa objavujú už v dojčenskom veku. Príčinou je mutácia v géne pre WAS proteín, ktorý je kľúčovým regulátorom bunkovej signalizácie a reorganizácie cytoskeletu hematopoetických buniek. Imunodeficiencia má charakter kombinovanej protilátkovej a celulárnej poruchy. Pacienti trpia bakteriálnymi infekciami horných a dolných dýchacích ciest, otitídami, ale aj oportunistickými infekciami a autoimunitnými komplikáciami. Zvýšená vnímavosť na malignity je dôsledkom poruchy NK aktivity a ďalších mechanizmov imunologického dohľadu. Novšie sa ukazuje, že WAS proteín zabezpečuje v ľudských bunkách genómovú stabilitu, ktorej porušenie vedie k lymforetikulárnym malignitám. Pred zavedením transplantácie krvotvorných buniek sa pacienti nedožívali dospelého veku v dôsledku krvácavých alebo infekčných komplikácií. Substitúcia imunoglobulínov a profylaktická antiinfekčná liečba umožňujú čiastočne znížiť infekčné komplikácie a pripraviť pacienta na čo najskoršiu transplantáciu. Vývoj malígnej komplikácie hrozí u 13 – 23 % pacientov (7). 90 % predstavujú leukémie, myelodysplázie alebo lymfómy. Závažnou komplikáciou je EBV infekcia. Podľa skorších prác bolo dvojročné prežívanie u pacientov s WAS a malignitou < 5% (8).
Na X chromozóm viazanú lymfoproliferatívnu chorobu (XLP) charakterizuje extrémne vysoká vnímavosť na EBV infekciu. Spôsobujú ju defekty v génoch s označením SH2D1A (XLP1) a XIAP/BIRC4 (XLP2). Porušená je NK aktivita a cytotoxická T lymfocytová imunita. Klinicky sa ochorenie manifestuje fulminantnou infekčnou mononukleózou s obrazom hemofagocytózy a zlyhania pečene. U pacientov, ktorí prekonajú mononukleózu, sa vyvíja malígny B lymfóm, hypogamaglobulinémia alebo lymfoproliferatívne ochorenie typu lymfoidnej vaskulitídy alebo malígneho NHL vysokého stupňa, najčastejšie s intestinálnym postihnutím. Priemerný vek manifestácie je okolo 6. roku života. Na toto ochorenie by sa malo myslieť vždy, keď sa u chlapca s lymfómom vyvíja po iniciálnej remisii druhý, odlišný typ lymfómu alebo hypogamaglobulinémia. V liečbe lymfómu sa používa štandardná chemoterapia a kuratívnou liečbou je alogénna transplantácia krvotvorných buniek. V predtransplantačnom období sa okrem substitučnej liečby používa v posledných rokoch rituximab.
Hyper-IgM syndróm (HIGM) môže mať niekoľko foriem, z ktorých každá postihuje v rôznej miere protilátkovú a celulárnu imunitu. X viazaná forma HIGM (X-HIGM) je dôsledkom porušenej expresie CD40 ligandu na aktivovaných T lymfocytoch. Príznaky sa manifestujú v prvých 2 rokoch života a prejavujú sa obrazom kombinovanej protilátkovej a T lymfocytovej imunodeficiencie. Pacienti majú veľmi nízke koncentrácie všetkých imunoglobulínov s výnimkou IgM. Postihnutí chlapci neprospievajú, majú neutropéniu, stomatitídu a sklerotizujúcu cholangitídu. Závažnou komplikáciou sú oportúnne infekcie ako pneumocystová pneumónia a bakteriálne infekcie gastrointestinálneho traktu. U pacientov sa v zvýšenej miere objavujú karcinómy pečene, pankreasu a žlčových ciest. V porovnaní s ostatnými PID sa u X-HIGM častejšie vyskytuje neuroendokrinný karcinóm.
Poruchy reparácie DNA
Viac než tretina malignít spojených s imunodeficienciou súvisí s poruchami obnovy (reparácie) DNA. Do tejto skupiny patrí niekoľko viac alebo menej známych zriedkavých chorôb. Najvyššie riziko malignity zo všetkých PID sa vyskytuje u ataxie telangiektázie (AT). Incidencia choroby sa uvádza 1 prípad na 100 000 narodených detí, ale mutácia v heterozygotnej forme sa dá identifikovať až u 2 % jedincov bežnej populácie (9). Vyskytuje sa u všetkých rás a rovnomerne medzi pohlaviami. Ataxia telangiektázie je autozomálne dominantná choroba, ktorú charakterizuje progresívna cerebelárna ataxia a dyzartria. Okulokutánne telangiektázie nie sú prítomné od narodenia, vyvíjajú sa postupne v priebehu prvej dekády života. V laboratórnom obraze je prítomná lymfopénia. Klinický a laboratórny nález býva veľmi variabilný, dokonca aj v rámci jednej rodiny. Typická je rádiosenzitivita so zvýšenou vnímavosťou buniek na ionizujúce žiarenie, najmä röntgenové a žiarenie gama alebo niektoré rádiomimetické chemické látky (vrátane farmák). Produkt ATM génu, ktorého mutácia je príčinou choroby, pôsobí za fyziologických okolností ako senzor zlomov dvojvláknovej DNA. Gén hrá významnú úlohu v genómovej stabilite, má pro-apoptotické účinky a dôležitú úlohu plní v procese diferenciácie kmeňových buniek. Porucha diferenciácie sa v tkanivách prejavuje vzostupom alfa-fetoproteínu, sérového proteínu produkovaného v pečeni. Sérové koncentrácie sú u pacientov extrémne vysoké a majú dôležitú diagnostickú hodnotu. Malígna transformácia buniek a tkanív sa vyskytuje až u 40 % pacientov s AT. Ide prevažne o leukémie a lymfómy. Toto riziko je 500-násobne vyššie oproti bežnej populácii. Vrcholným obdobím je adolescencia, hoci opísané sú aj prípady v dojčenskom období, dokonca pred stanovením diagnózy AT. Z tohto dôvodu je v rámci diferenciálnej diagnostiky u lymfoidných malignít v mladšom veku nutné myslieť na možnosť AT. Druhou častou formou malígneho ochorenia u AT sú epitelové karcinómy. Veľmi často bývajú viacpočetné.
Z hľadiska onkologickej liečby je AT komplikovaná, pretože pacienti sú veľmi senzitívni na radiáciu a bežné dávky žiarenia ťažko poškodzujú tkanivá. Z tohto dôvodu nie je možné použiť rádioterapiu. Pacienti ale obvykle tolerujú štandardné chemoterapeutické režimy. Nevhodné sú alkylačné látky a obmedzené použitie má metotrexát. Zvýšená rádiosenzitivita existuje taktiež u zdravých nosičov. ATM gén ovplyvňuje známe tumor supresorické gény ako sú TP53 a BRCA1, ktoré sú spojené s predispozíciou k rakovine prsníka. Zdravé ženy, nosičky mutácie v ATM géne (heterozygotná mutácia), majú zvýšené riziko rakoviny a riziko sa ešte zvyšuje u fajčiarok. Aj v tomto prípade je otázne využitie rádioterapie.
Veľmi podobným syndrómom ako AT je Nijmegenský zlomový syndróm (NBS). Toto autozomálne recesívne ochorenie sa častejšie objavuje u slovanskej populácie. NBS gén kóduje nibrín (p95), ktorý je jedným z komponentov multiproteínového komplexu a súčasťou identickej dráhy ako produkty ATM génu. NBS má preto veľmi podobné charakteristiky ako AT vrátane zvýšenej vnímavosti na chromozómové zlomy indukované radiáciou. Z klinického hľadiska sa u pacientov vyvíja kombinovaná imunodeficiencia so závažnou poruchou tvorby protilátok (agamaglobulinémia), lymfopéniou (deficitom CD4+ aj CD8+ T lymfocytov), chromozómovou instabilitou a predispozíciou k malignitám. Súčasne majú postihnutí jedinci typické dysmorfné črty s mikrocefaliou a hypoplastickým neurokrániom. Lymfóm ako najčastejšia malignita sa vyvíja často už v predškolskom období. Podobne ako v prípade AT nie je možné použitie rádioterapie. V liečbe sa využívajú redukované chemoterapeutické postupy.
Poruchy fagocytózy
Kvantitatívne a funkčné poruchy fagocytových funkcií predstavujú skupinu viac než dvoch desiatok klinických jednotiek. K najzávažnejším patrí ťažká kongenitálna neutropénia. Ide o heterogénnu poruchu vyzrievania myeloidnej vývojovej rady na úrovni stupňa promyelocyt – myelocyt. Absolútny počet neutrofilov v periférnej krvi sa pohybuje pod 0,5x10exp9/L. Klinicky sú prítomné ťažké bakteriálne infekcie už v dojčenskom veku. Najčastejším genetickým defektom je mutácia génu ELANE (neutrofilová elastáza), čo je serínová proteáza hydrolyzujúca mnohé proteíny v lyzozómoch (azurofilných granulách) a extracelulárnom matrixe. Súčasne je schopná poškodzovať proteíny vonkajšej membrány E. coli a ďalších enteropatogénnych baktérií. Miernejšia forma sa manifestuje obrazom cyklickej neutropénie. Klinický obraz kongenitálnej neutropénie majú aj defekty v génoch HAX1 a G6PC3. V liečbe sa používa denná aplikácia kolónie stimulujúceho faktora granulocytov (G-CSF), ktorá zvyšuje počet neutrofilov a minimalizuje infekčné komplikácie. Kongenitálna neutropénia sa spája s častým výskytom leukémií, prevažne akútnej myeloidnej leukémie. Uvádza sa ale aj akútna lymfoidná leukémia, chronická myelomonocytová leukémia a bifenotypická leukémia. Medzi dávkou G-CSF a výskytom leukémie existuje priama úmera. U pacientov, u ktorých sa leukémia vyvinula, sa potvrdili bodové mutácie génu pre G-CSFR (receptor), ktoré sa u primárnej akútnej myeloidnej leukémie nevyskytujú (10).
Liečba a prognóza
Prognóza pacientov s malignitou a imunodeficienciou je obvykle horšia v porovnaní s imunokompetentnými pacientmi. Imunodeficientní pacienti majú malígne ochorenia častejšie diseminované a v nižšom stupni zrelosti. Cytotoxickú liečbu tolerujú horšie a je spojená s podstatne vyšším rizikom infekčných komplikácií a orgánovým poškodením. Na druhej strane, na liečbu neexistuje primárna rezistencia a pacienti môžu byť úspešne liečení. Imunodeficientní pacienti vyžadujú individuálny prístup podľa typu imunitnej poruchy a aktuálneho imunitného statusu. Nevyhnutná je agresívnejšia liečba infekcií, najmä profylaxia pneumónie vyvolanej Pneumocystis jirovecii. Štandardnou súčasťou liečby je substitúcia IgG.
V posledných rokoch sa rozširujú indikácie pre transplantáciu krvotvorných buniek. Pre pacientov, ktorí prekonali lymfoproliferatívne ochorenie, to môže byť prognosticky najlepšie riešenie. Prípravné režimy s redukovanou intenzitou sa ukázali ako úspešná alternatíva u pacientov s WAS, SCID a v niektorých ďalších prípadoch. Prognózu zlepšujú aj nové biologiká. Rituximab (anti-CD20) sa úspešne použil v iniciálnej liečbe lymfoproliferatívnej choroby a v supresii EBV infekcie. Prvé úspechy zaznamenáva – v prípade pacientov s ťažkou kombinovanou imunodeficienciou – génová terapia.
Záver
Kauzálny vzťah medzi poruchou imunity a onkologickým procesom je obojsmerný. Imunodeficiencia/imunosupresia sa môže vyvinúť u imunokompetentných pacientov s malignitou sekundárne, ako dôsledok samotnej malignity alebo jej liečby (chemoterapia, transplantácia). Podobne ako v prípade pacientov s PID je nevyhnutné zvládnuť infekčné komplikácie. Viaceré štúdie potvrdili, že aplikácia imunoglobulínov u pacientov s chronickou lymfocytárnou leukémiou a u pacientov s NHL viedla k poklesu bakteriálnych infekcií. Lymfóm alebo iné malígne ochorenie býva zriedkavo prvým príznakom PID. Hematológovia a onkológovia by nemali zabúdať na takúto možnosť, najmä u detských pacientov. Posúdenie imunologického statusu je dôležitou súčasťou diagnostického a terapeutického procesu u onkologického pacienta.
Literatúra
- Picard C, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, Etzioni A, Holland SM, Klein C, Nonoyama S, Ochs HD, Oksenhendler E, Puck JM, Sullivan KE, Tang ML, Franco JL, Gaspar HB. Primary Immunodeficiency Diseases: an Update on the Classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J Clin Immunol. 2015 Oct 19.
- The French national registry of primary immunodeficiency diseases CEREDIH: The French PID study group. Clinical Immunology (2010) 135, 264 – 272.
- Gross TG, Shiramizu B.Lymphoproliferative disorders and malignancies related to immunodeficiencies. In: Pizzo PA,Poplack DG, editors. Principles and Practice of Pediatric Oncology. Philadelphia, PA, Lippincott Williams & Wilkins (2006) 748 – 767.
- Salavoura K, Koliazlexi A, Tsangaris G, Mavrou A. Development of cancer in patients with primary immunodeficiencies. Anticancer Res (2008) 28, 1263 – 1270.
- Vajdic CM, Mao L, van Leeuwen MT, et al. Are antibody deficiency disorders associated with a narrower range of cancers than other forms of immunodeficiency? Blood (2010) 116, 1228 – 1234.
- Cunningham-Rundles C, Siegal FP, Cunningham-Rundles S, Lieberman P. Incidence of cancer in 98 patients with common varied immunodeficiency. J Clin Immunol (1987) 7, 294 – 299.
- Bosticardo M, Marangoni F, Aiuti A. a spol. Recent advances in understanding the pathophysiology of Wiskott-Aldrich syndrome. Blood (2009) 113, 6288 – 6295.
- Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr (1994) 125, 876 – 885.
- Swift M, Morrell D, Cromartie E, Chamberlin AR, Skolnick MH, Bishop DT. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am J Hum Genet. (1986) 39 (5), 573–583.
- Germeshausen M, Ballmaier M, Welte K. Incidence of CSF3R mutations in severe congenital neutropenia and relevance for leukemogenesis: Results of a long-term survey. Blood (2007) 109, 93 – 99.




