

Familiárna hypercholesterolémia – jedno z najčastejších autozómovo dominantne dedičných ochorení – je charakterizovaná zvýšenou hladinou LDL cholesterolu, akcelerovanou aterosklerózou a predčasnou manifestáciou kardiovaskulárnych ochorení. Veľké množstvo pacientov s týmto ochorením o svojom postihnutí nevie a nie sú adekvátne liečení. Cieľom projektu MedPed FH je aktívne vyhľadávanie pacientov s familiárnou hypercholesterolémiou, kaskádový skríning u ich rodinných príslušníkov a následná adekvátna liečba a predchádzanie predčasnej manifestácie kardiovaskulárnych ochorení.
Familiárna hypercholesterolémia (FH) je najčastejšie geneticky podmienené ochorenie spôsobujúce predčasné kardiovaskulárne ochorenie a smrť. Toto autozómovo dominantne dedičné ochorenie je charakterizované zvýšenou hladinou LDL cholesterolu (LDL-C), akcelerovanou aterosklerózou a predčasnou manifestáciou kardiovaskulárnych ochorení (Goldstein, 1995). Ako prvý opísal familiárnu hypercholesterolémiu nórsky lekár Carl Müller v tridsiatych rokoch minulého storočia ako ochorenie charakterizované šľachovou xantomatózou, vysokou hladinou cholesterolu, infarktom myokardu v mladom veku a familiárnym výskytom (Müller, 1938).

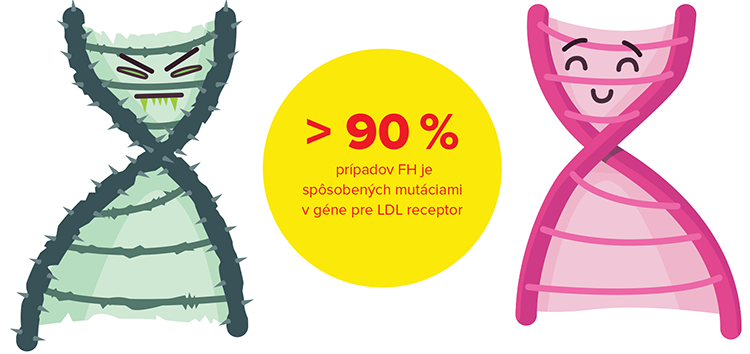
Názov familiárna hypercholesterolémia sa používa spoločne pre autozómovo dominantne podmienené hypercholesterolémie spôsobené mutáciami v súčasnosti troch známych génov: génu pre LDL receptor (tzv. LDLR mutácie), génu pre apolipoproteín B (mutácia Arg3500Gln, pre ktorú sa používa aj názov familiárny defektný apoB-100 – FDB) a génu pre proproteín konvertázu subtilizín/kexín 9 (PCSK9) (Nordestgaard, 2013). Viac než 90 % prípadov FH je však spôsobených mutáciami v géne pre LDL receptor, pričom doteraz bolo identifikovaných > 1 200 rôznych mutácií génu pre LDLR. Zastúpenie FDB v populácii pacientov s klinickou diagnózou FH je v rámci Európskych krajín veľmi rôznorodé, v rozmedzí 0 – 11 %, pričom 9,7 % zistených na Slovensku je porovnateľných s údajmi z Českej republiky a Poľska (Gašparovič, 2007). Mutácie v géne pre PCSK9 sa vyskytujú u menej než 1 % pacientov s FH.
Aj keď sa mutácie všetkých troch génov prejavujú obdobným klinickým obrazom (vysoké LDL, predčasné kardiovaskulárne ochorenie, prítomnosť šľachových xantómov), existuje určitá variabilita klinického fenotypu, jednak medzi jednotlivými génmi, ale aj medzi rôznymi mutáciami rovnakého génu. V niektorých populáciách FH mali pacienti s geneticky preukázaným FDB menej závažný klinický fenotyp než ostatní FH pacienti (Vohnout, 2003; Nordestgaard, 2013). Zaznamenané priemerné hladiny LDL cholesterolu u pacientov s FH však môžu byť významne ovplyvnené aj rôznymi klinickými kritériami, na základe ktorých je stanovená klinická diagnóza FH a indikované prípadné DNA vyšetrenie (Vohnout, 2001). V nezanedbateľnom množstve klinicky diagnostikovaných pacientov s FH sa neidentifikuje žiadna z doteraz objavených mutácií, čo vedie k úvahám o ďalších potenciálnych génoch zodpovedných za FH. Okrem spomenutých autozomálne dominantných foriem FH bola v nedávnom období identifkovaná aj autozomálne recesívna forma FH (Zuliani, 1999).
Klinická prezentácia a výskyt FH

Pri heterozygotnej forme familiárnej hypercholesterolémie dedí postihnutý jedinec jeden defektný gén od jedného rodiča a druhý, normálny, od druhého rodiča. To vedie k nedostatočnému metabolizovaniu LDL a približne dvojnásobne zvýšenej hladine LDL cholesterolu v plazme. Celkový cholesterol u neliečených dospelých sa zvyčajne pohybuje v rozmedzí 8 až 15 mmol/l, LDL cholesterol 5 – 10 mmol/l so zvyčajne normálnou hladinou triacylglycerolov a normálnou alebo zníženou hladinou HDL cholesterolu (Hovingh, 2013; Nordestgaard, 2013; Rašlová, 2009).
Heterozygotná forma FH (heFH) sa vyskytuje vo väčšine populácií vo frekvencii 1 : 200 – 500 (Nordestgaard, 2013). Vzhľadom na autozómovo dominantný typ prenosu je pravdepodobnosť postihnutia prvostupňových príbuzných pacienta s heFH 50 %, pravdepodobnosť pre druhostupňových a treťostupňových je 25 % a 12,5 %. Okrem zvýšenej plazmatickej hladiny cholesterolu sa môžu vyskytovať aj viditeľné znaky cholesterolových depozitov, ako sú arcus lipoides corneae, xanthelasma palpebrarum, ale hlavne šľachové xantómy, ktoré sú patognomickým znakom FH, jeho neprítomnosť však FH nevylučuje. Predilekčnými miestami výskytu šľachových xantómov sú Achillove šľachy, šľachy extenzorov rúk a lakťov (Thompson, 1999). Naša empirická skúsenosť však poukazuje na ich relatívne nižší výskyt oproti údajom zo staršej literatúry, túto skúsenosť potvrdzujú aj údaje z Českej republiky, kde zaznamenali výskyt šľachových xantómov len u 2,4 % FH pacientov vo veku do 45 rokov a u 13,3 % pacientov vo veku nad 45 rokov (Freiberger, Češka, 2007). Vzhľadom na relatívne častý výskyt korneálneho arcusu vo vyšších vekových skupinách aj vo všeobecnej populácii pokladáme takýto nález za suspektný pre výskyt FH len u mladších pacientov. Obdobne nález xanthelasma palpebrarum nie je pre FH špecifický, tento nález zvažujeme v súvislosti s FH len u mladých pacientov.
Klinicky manifestná ischemická choroba srdca (ICHS) sa zvyčajne vyskytuje v priemernom veku 45 – 48 rokov u mužov a vo veku 55 – 58 rokov u žien (WHO, 1999). Riziko prekonania IM vo veku do 30 rokov u neliečeného muža s heFH je asi 5-percentné, do 50 rokov 50-percentné a vo veku do 60 rokov 85-percentné. U žien je toto riziko 1-percetné do 30 rokov, 15-percentné do 50 rokov a 50-percentné do 60 rokov (Illingworth, 1995). Prevalencia ICHS u osôb s FH z nedávno publikovanej veľkej populačnej dánskej štúdie bola 33 %, pričom polovica pacientov s FH nebola liečená statínom (Benn, 2012). Riziko ICHS u tých FH pacientov, ktorí neboli liečení statínom, bolo 13-krát vyššie v porovnaní s osobami bez FH. Pacienti s FH liečení statínom mali toto riziko 10-krát vyššie, čo svedčí o nedostatočnej dávke statínu, respektíve o neskorom začatí takejto liečby. Toto potvrdzujú aj výsledky sledovaní z viacerých krajín, ktoré ukázali, že veľké množstvo pacientov s FH nevie o svojom ochorení a aj tí, u ktorých bola diagnóza FH stanovená, často nie sú adekvátne liečení (Nordestgaard, 2013).
Keďže hladiny LDL cholesterolu sú zvýšené od narodenia, pacient s FH je celoživotne exponovaný týmto hlavným rizikovým faktorom aterosklerózy, následkom čoho je veľmi vysoké celoživotné riziko kardiovaskulárnych ochorení. Preto aj súčasné odporúčania Európskej kardiologickej spoločnosti (ESC) a Európskej aterosklerotickej spoločnosti (EAS) zaraďujú pacientov s FH automaticky do skupiny s vysokým kardiovaskulárnym rizikom, bez ohľadu na prítomnosť ostatných rizikových faktorov a vypočítané SCORE riziko (Perk, 2012; Reiner, 2011).
Pri homozygotnej forme familiárnej hypercholesterolémie dedí postihnutý jedinec defektný gén od oboch rodičov, preto je aj postihnutie ťažšie. Celkový cholesterol u postihnutého nadobúda hodnoty vyššie než 15mmol/l, nezriedka aj okolo 30mmol/l. LDL cholesterol je asi 4 – 5-násobne vyšší než u zdravého jedinca. ICHS sa u týchto pacientov manifestuje v útlom detstve a bez liečby zomierajú na kardiovaskulárnu príhodu do dvadsiateho roku života. Prevalencia homozygotnej formy FH je približne 1 : 1 000 000 (Nordestgaard, 2013).
Diagnostika FH

Kritériá používané na identifikáciu FH zahŕňajú biochemické ukazovatele (celkový a LDL cholesterol), prítomnosť šľachových xantómov a rodinnú alebo osobnú anamnézu predčasného výskytu ICHS. Všeobecne akceptované a využívané sú tri diagnostické schémy. US MedPed kritériá využívajú pre určenie diagnózy FH hladinu cholesterolu v závislosti od veku a stupňa príbuznosti k probandovi (Tabuľka č. 1). Holandské kritériá určujú diagnózu na základe zhodnotenia širšieho spektra znakov, ktoré zahŕňajú preverenie rodinnej záťaže, osobnej anamnézy, vyšetrenie LDL cholesterolu pacienta i jeho fyzikálne vyšetrenie a DNA analýzu. Výsledok každej kategórie sa oboduje a na základe konečného bodového súčtu sa určuje definitívna, pravdepodobná alebo možná diagnóza (Tabuľka č. 2). Takzvané Simon Broome diagnostické kritériá (Tabuľka č. 3) hodnotia hladinu celkového a LDL cholesterolu, prítomnosť šľachových xantómov u prvostupňových aj druhostupňových príbuzných, pozitívnu rodinnú anamnézu a dôkaz mutácie spôsobujúcej FH.


V bežnej klinickej praxi treba zvážiť diagnózu FH u pacienta, ktorý spĺňa tieto kritériá výskytu. Hladiny celkového cholesterolu nad 7,5 mmol/l alebo LDL cholesterolu nad 4,9 mmol/l (dospelí pacienti), respektíve celkový cholesterol nad 6,7 mmol/l alebo LDL cholesterol nad 4,0 mmol/l u detí do 16 rokov a zároveň spĺňa aspoň jeden z ďalších uvedených parametrov:
- infarkt myokardu pod 65 rokov u prvostupňových príbuzných a pod 50 rokov u druhostupňových príbuzných,
- prítomnosť zvýšenej hladiny celkového (nad 7,5 mmol/l u dospelých, nad 6,7 mmol/l do veku 16 rokov) alebo LDL cholesterolu (nad 4,9 mmol/l u dospelých, nad 4,0 mmol/l do veku 16 rokov) u prvostupňových aj druhostupňových príbuzných,
- nález šľachových xantómov u pacienta alebo jeho prvostupňových (rodič, súrodenci, deti) alebo druhostupňových (starí rodičia, strýko, teta) príbuzných.
V prípade identifikácie takéhoto pacienta je vhodné odoslať pacienta na špecializované pracovisko, ktoré zabezpečí v rámci projektu MedPed FH vyšetrenie príbuzných pacienta a prípadne aj genetické vyšetrenie. Zoznam pracovísk, ktoré sa venujú tejto problematike možno nájsť na stránke MedPed projektu (www.medpedfh.sk).
Liečba pacienta s FH vyžaduje zvyčajne vysoké dávky potentných statínov, často aj v kombinácii s ezetimibom. Aj napriek takejto liečbe však u časti pacientov nie je možné dosiahnuť cieľové hladiny LDL cholesterolu. V blízkej budúcnosti očakávame príchod novej skupiny potentných hypolipidemík – tzv. inhibítory PCSK9, ktoré sa podľa výsledkov doposiaľ realizovaných klinických štúdií javia ako veľmi vhodná terapia pre pacientov s FH.
Projekt MedPed FH na Slovensku
Vyhľadávaniu a liečbe pacientov s FH sa celosvetovo venuje projekt MedPed FH (Make Early Diagnosis – Prevent Early Deaths in Medical Pedigrees), ktorý funguje už viac než desaťročie aj na Slovensku. Projekt vznikol v 90. rokoch minulého storočia z iniciatívy profesora Williamsa z Utahu s cieľom zlepšiť diagnostiku a liečbu pacientov s FH a znížiť počet úmrtí na kardiovaskulárne ochorenia v rodinách s FH. Vyhľadávanie pacientov v tomto projekte prebieha metódou kaskádového skríningu v rodinách probanda, čo umožňuje ekonomicky najefektívnejšie vyhľadávanie postihnutých jedincov a ich následnú liečbu. Ide o spoluprácu medzi pacientmi, ich rodinami a zdravotníkmi, ako sú lekári prvého kontaktu, špecialisti, nutriční špecialisti, sestry a predstavitelia verejného zdravotníctva. Podstatou projektu je zavedenie účinného systému, ktorý zabezpečí vyšetrenie osôb s vysokým rizikom FH, keďže sú príbuzní s pacientom, ktorý má FH, pričom tieto osoby dostanú informáciu o riziku, sú edukované a dispenzarizované.
MedPed postup je pre FH vhodný z viacerých dôvodov (Williams, 1993)
- FH má relatívne vysokú prevalenciu, pričom metabolická porucha spôsobuje, že títo pacienti majú veľmi frekventovanú včasnú aterosklerózu, a teda predstavujú závažný zdravotnícky problém.
- Hĺbka a šírka vedomostí z genetiky, molekulárnej biológie, charakteristika a klinické následky FH poskytujú významný vedecký materiál a dôvody pre MedPed FH.
- Relatívne dlhá latentná perióda do manifestácie klinických následkov FH poskytuje dostatok času na stanovenie diagnózy a zavedenie preventívnych postupov.
- Dostupnosť odborníkov a vhodných diagnostických testov, ktoré obvykle zahŕňajú odber krvi na biochemické a/alebo genetické vyšetrenia.
- Dostupnosť akceptovateľných a efektívnych intervenčných stratégií založených na dôkazoch.
- Možnosti podpory a informovania pacientov, rodiny a lekárov prvého kontaktu.
- MedPed FH vytvára trvalý register postihnutých osôb, ktorý poskytuje dôležité zdravotnícke a medicínske informácie.
Stratégia vyhľadávania pacientov postihnutých FH v rizikových rodinách (kaskádový skríning) je ekonomicky, ale aj prakticky najefektívnejším spôsobom vyhľadávania postihnutých jedincov a ich následnej liečby. Po stanovení diagnózy FH u probanda (prvý člen rodiny so zisteným ochorením) treba zabezpečiť vyšetrenie lipidového spektra u všetkých prvostupňových príbuzných (riziko FH u nich je 50 %). Následne u tých príbuzných, u ktorých FH potvrdíme, treba zabezpečiť vyšetrenie všetkých jeho prvostupňových príbuzných. V bežnej klinickej praxi môže takýto postup prinášať rôzne komplikácie (časová náročnosť, problémy s poisťovňami), preto je vhodné odoslať pacienta so suspektnou diagnózou FH na niektoré špecializované pracovisko MedPed FH, ktoré je schopné zabezpečiť komplexnú starostlivosť o pacienta, vyšetrenie príbuzných a ich adekvátnu liečbu. V projekte MedPed je v spolupráci s laboratóriom diabetu a porúch metabolizmu (DIABGENE) Ústavu experimentálnej endokrinológie SAV v rámci vedeckých grantov zabezpečená aj DNA diagnostika tohto ochorenia, ktorá sa stáva diagnostickým štandardom.
Na Slovensku je do projektu MedPed FH v súčasnosti zapojených 14 špecializovaných pracovísk, na ktoré je možné odoslať pacientov s FH alebo suspektnou FH. Koordináciu projektu zabezpečuje koordinačné centrum pre familiárne hyperlipoproteinémie na Slovenskej zdravotníckej univerzite v Bratislave. Kontakt na jednotlivé centrá a koordinačné centrum možno nájsť na www.medpedfh.sk. Uvedená stránka poskytuje okrem kontaktov aj podrobnejšie informácie o problematike FH a projekte MedPed FH a edukačných podujatiach pre lekárov.
Záver
Napriek vysokému riziku vzniku predčasných kardiovaskulárnych príhod ostáva FH významne poddiagnostikovaná, vysoké riziko pacientov s FH je často prehliadané, alebo nesprávne klasifikované a títo pacienti sú nedostatočne liečení. Ambíciou projektu MedPed FH na Slovensku je zlepšiť identifikáciu pacientov s FH, ich aktívne vyhľadávanie, adekvátnu liečbu a predchádzať predčasným kardiovaskulárnym komplikáciám. Toto úsilie však vyžaduje zapojenie a spoluprácu širokej odbornej verejnosti.
Literatúra
- Benn M., Watts G. F., Tybjaerg-Hansen A., Nordestgaard B. G. Familial Hypercholesterolemia in the Danish General Population: Prevalence, Coronary Artery Disease, and Cholesterol-Lowering Medication. J Clin Endocrinol Metab 2012; 97: 3956 – 64
- ESC/EAS Guidelines for the management of dyslipidaemias. Eur Heart J 2011; 32, 1769 – 1818
- Freiberger T., Češka R. Je familiární hypercholesterolemie v České republice pod kontrolou? Vnitr. Lék. 2007; 53: 703 – 708
- Gašparovič J., Bašistová Z., Fábryová Ľ., Wsólová L., Vohnout B., Rašlová K. Familial Defective Apolipoprotein B-100 in Slovakia. Are Differences in Prevalence of FDB Explained by Ethnicity? Atherosclerosis 2007; 194(2): e95 – 107
- Goldstein J. L., Hobbs H. H., Brown M. S. Familial hypercholesterolaemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D (editors). The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 1995. pp. 1981 – 2030
- heartuk.org.uk: http://www.heartuk.org.uk/HealthProfessionals/downloads/05_advice_and_information/HUK_AS04_Diagnostic.pdf
- Hovingh G. K., Davidson M. D., Kastelein J. J., O’Connor A. M. Diagnosis and treatment of familial hypercholesterolaemia. Eur Heart J. 2013; 34: 962 – 971
- Illingworth D. R., Duell P. B., Connor W. E. Disorders of lipid metabolism. Endocrinology and Metabolism 1995, pp. 1315 – 1405
- Müller C. Xanthomata, hypercholesterolaemia, angina pectoris. Acta Med Scand 1938; 89:75 – 80
- Nordestgaard B. G., Chapman M. J., Humphries S. E., et al; for the European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. Eur Heart J. 2013 Dec; 34 (45):3478 – 90a
- Perk J., et al. European Guidelines on cardiovascular disease prevention in clinical practice (version 2012). European Heart Journal. 2012; 33: 1635 – 1701
- Rašlová K., Ivaničová M. Familiárna hypercholesterolémia a prevencia jej následkov. WHO MEDPED projekt. Cardiol 2000; 9(1): 19 – 23
- Reiner Z., et al. ESC/EAS Guidelines for the management of dislipidaemias. European Heart Journal., 2011; 32:1769 – 1818
- Thompson G. R. Familial hypercholesterolemia. In: Betteridge D. J, Illingworth D. R, Shepherd J. Lipoproteins in health and disease. London: 1999; 675 – 692
- Vohnout B., Rašlová K., Gašparovič J., Donati M. B., Iacoviello L. Familial hypercholesterolaemia. Lancet. 2001; 357:1712
- Vohnout B, Raslova K, Gasparovic J, Franekova J, Fabryova L, Belosovicova M, Kovac G, Sebova C, Rajecova E, Stavny J, Babjak M, Donati MB, Iacoviello L. Lipid levels and their genetic regulation in patients with familial hypercholesterolemia and familial defective apolipoprotein B-100: the MEDPED Slovakia Project. Atherosclerosis Suppl. 2003; 4:3 – 5
- Williams et al. Am J Cardiology 1993; 72(2):171 – 6
- Williams R. R., Schumaker C., Barlow G., et al. Documented need for more effective diagnosis and treatment of familial hypercholesterolaemia according to data from 502 heterozygotes in Utah. Am J Cardiol 1993:72:18D – 24D
- World Health Organization: Familial hypercholesterolemia – report of a second WHO Consultation. Geneva, Switzerland: World Health Organization 1999 (WHO publication No. WHO/HGN/FH/CONS/99.2)
- Zuliani G., Arca M., et al. Characterization of a new form of inherited hypercholesterolemia: familial recessive hypercholesterolemia. Arterioscl Thromb Vasc Biol 1999; 19: 802 – 809

