







Štandardy v starostlivosti o pacienta s myelodysplastickým syndrómom (MDS) sa neustále menia. Pacient s MDS by mal byť v starostlivosti hematológa, pretože on vie najlepšie posúdiť súčasné terapeutické možnosti, ako aj možnosť realizácie transplantácie kostnej drene. Liečebná stratégia je založená na novelizovanom medzinárodnom prognostickom skórovacom systéme IPSS-R. Pacienti dostávajú v súčasnosti hlavne podpornú liečbu, ktorá na jednej strane predlžuje celkové prežívanie a zlepšuje kvalitu života, ale na druhej strane ide len o dočasné opatrenia. Z hľadiska budúcnosti treba nájsť také terapeutické možnosti, ktoré by stimulovali produkciu v kostnej dreni dlhodobo.
Definícia
Myelodysplastický syndróm (MDS) je označenie pre heterogénnu skupinu ochorení s klonálnou hemopoézou, ktoré sa vyznačujú aberantnou myeloidnou diferenciáciou, dysplastickými zmenami, neefektívnou krvotvorbou a rastúcou genomickou nestabilitou, ktoré sa klinicky prejavia periférnou cytopéniou (1).
Epidemiológia
V roku 1995 bola odhadovaná ročná miera incidencie MDS v USA 1 500 prípadov (2). Dve novšie štúdie, ktoré na výpočet využívali už novozavedený register nádorových ochorení v USA, odhadli ročnú mieru výskytu na > 10 000 (3, 4). Štúdia z roku 2003 predpokladá približne 45 000 nových prípadov MDS v USA u ľudí ≥ 65 rokov (5). A úplne posledná štúdia z USA odhadla ročnú incidenciu na 75 prípadov zo 100 000 obyvateľov ≥ 65 rokov (6). Žiaľ, údaje výskytu MDS na Slovensku momentálne nie sú dostupné. Existujúce údaje konzistentne ukazujú, že MDS je prevažne ochorenie starších ľudí. Približne 86 % pacientov s MDS je v čase stanovenia diagnózy vo veku ≥ 60 rokov (medián veku je 76 rokov) a iba 6 % prípadov bolo diagnostikovaných u osôb vo veku ≤ 50 rokov (5). Muži majú vyššiu frekvenciu výskytu než ženy a biela rasa má vyššiu frekvenciu výskytu než iné rasové/etnické skupiny (3). So starnúcim obyvateľstvom rastie aj počet prípadov MDS.
Etiológia
Vrodené ochorenia krvotvorby, ako napríklad Fanconiho anémia, zvyšujú riziko rozvoja MDS (7). Okrem toho, ionizujúce žiarenie a chemoterapia pre predchádzajúcu malignitu, ako aj expozícia benzénom sú považované za ďalšie rizikové faktory (8). Pacienti so sekundárnym MDS majú navyše oveľa horšiu prognózu. Hlavným zdrojom benzénu je vo všeobecnosti cigaretový dym (9). K environmentálnym rizikovým faktorom spojeným s MDS patrí fajčenie cigariet (10 – 12) a expozícia voči rozpúšťadlám a pesticídom (13 – 14). Vzťah medzi konzumáciou alkoholu a MDS bol posudzovaný v niekoľkých štúdiách. Výsledky týchto štúdií sú však protichodné. Niektoré štúdie ukázali, že spotreba alkoholu nemá vplyv na riziko rozvoja MDS (15 – 17). Iná štúdia naopak ukázala významnú súvislosť medzi konzumáciou alkoholu a MDS (18). Iná štúdia poukázala na významný vzťah medzi indexom telesnej hmotnosti (BMI) a následným vývojom MDS (17). Pacienti s nadváhou (BMI = 25 – 29,9) a obézni pacienti (BMI ≥ 30) mali vyššie riziko rozvoja MDS než jedinci s normálnou hmotnosťou.
Patogenéza
Za posledné roky sa vďaka novým technikám analýzy DNA zmenil pohľad na patogenézu MDS. Dlho sa totiž verilo, že génové mutácie sa pri MDS takmer vôbec nevyskytujú, s výnimkou MDS pacientov s vysokým rizikom progresie do akútnej myeloblastovej leukémie (AML). Väčšina týchto genetických zmien však nie je špecifická len pre MDS a vyskytuje sa aj pri iných myeloidných ochoreniach (18).
MDS je ochorenie hemopoetických kmeňových buniek. Genetické abnormality, ktoré sú zistené v zrelých myeloidných bunkách sú už prítomné aj v kmeňových bunkách (19). Nedávne práce ukázali, že rôzne mutácie môžeme nájsť v kmeňových bunkách. Tieto mutácie im dajú proliferačnú výhodu oproti normálnym kmeňovým bunkám bez toho, aby došlo k ovplyvneniu potenciálu progenitorových buniek (20). Prítomnosť určitých mutácií pri MDS má prognostický význam. Bejar et al. (21) venovali pozornosť mutáciám, ktoré sú spojené s nepriaznivým liečebným výsledkom a ich prítomnosť zvyšuje riziko progresie MDS do AML. Celkovo popísali mutácie piatich génov (TP53, EZH2, ETV6, RUNX1 a ASXL1), ktorých prítomnosť sa spája s kratším celkovým prežívaním.
Najčastejšou cytogenetickou abnormalitou u pacientov s MDS je parciálna alebo kompletná delécia dlhého ramienka chromozómu 5 del(5q). Vyskytuje sa u 10 – 15 % pacientov s de novo MDS (22). 5q mínus syndróm reprezentuje samostatný subtyp MDS podľa kritérií Svetovej zdravotníckej organizácie (WHO). Je asociovaný s nízkym rizikom transformácie do AML. Typicky postihuje ženy. Charakteristický je makrocytárnou anémiou, hypolobulizovanými megakaryocytmi, normálnym alebo zvýšeným počtom trombocytov, ľahkou neutropéniou a počet blastov v kostnej dreni je < 5 % (23).
V patogenéze MDS zohrávajú kľúčovú úlohu epigenetické zmeny. Patrí sem predovšetkým metylácia DNA. K epigenetickým zmenám nedochádza len na úrovni DNA. Zahrnuté sú aj posttranslačné zmeny proteínov, ktoré tvoria históny (acetylácia, metylácia a ubikvitácia) (24 – 26).
Klasifikácia
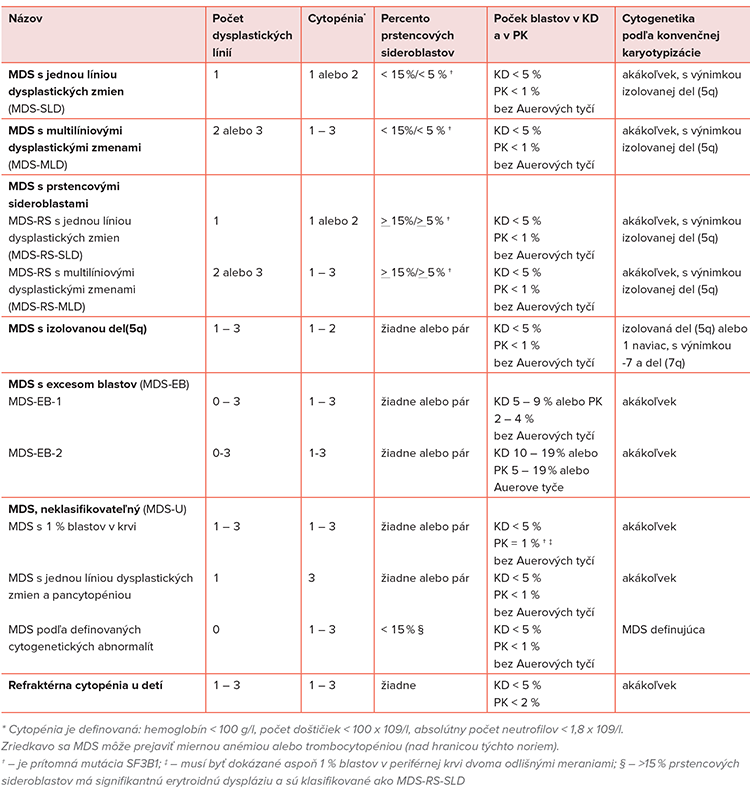
Dlhé roky sa v klasifikácii MDS používa francúzsko-americko-britská klasifikácia (FAB) z roku 1982, ktorá definovala 5 hlavných podtypov MDS: refraktérna anémia (RA), refraktérna anémia s excesom blastov (RAEB), refraktérna anémia s excesom blastov v transformácii (RAEBt), refraktérna anémia s prstencovými sideroblastami (RARS) a chronická myelomonocytová leukémia (CMMoL). FAB klasifikácia bola revidovaná Svetovou zdravotníckou organizáciou, posledná aktualizácia je z tohto roku (Tabuľka č. 1) (27).
Príznaky
Najčastejším príznakom MDS je anémia a s ňou súvisiaci anemický syndróm, ktorý je pozorovaný v 90 % prípadov. Asi 25 – 30 % pacientov má leukopéniu. Len asi 10 % pacientov má v čase stanovenia diagnózy trombocytopéniu. Symptómy vyplývajúce z porušenej hemopoézy tak môžu byť veľmi pestré. Začiatok ochorenia býva pomalý. Dôsledkom cytopénie bývajú časté infekcie s vysokou teplotou, krvácavé prejavy (petechie, sufúzie, hematómy, epistaxy), pacient udáva celkovú slabosť a únavu. V krvnom obraze je najčastejšie makrocytová anémia s ovalocytózou, anizocytózou a so známkami nezrelosti erytrocytov. Počet retikulocytov je zvyčajne veľmi nízky. Kostná dreň je v úvode hyperplastická (s výnimkou hypoplastických foriem MDS) s megaloblastovou prestavbou erytropoézy a s početnými dysplastickými zmenami vo všetkých vývojových radoch (28). Základom stanovenia diagnózy je vyšetrenie kostnej drene (cytologické, histologické, imunohistochemické a fenotypové). Dôležité je aj jej cytogenetické vyšetrenie, pretože, ako už bolo spomenuté, je častý nález chromozómových aberácií.
Prognóza
MDS zahŕňa skupinu ochorení s variabilným klinickým priebehom a s odlišným biologickým správaním. Časť pacientov je ohrozená progresiou do akútnej leukémie, iná časť pacientov sa stáva závislou na transfúziách, iná je náchylná na infekcie a krvácavé prejavy. U niektorých pacientov dochádza k okamžitej transformácii do akútnej leukémie, zatiaľ čo ostatní prežívajú dlhodobo a MDS má chronický priebeh. Preto bol vypracovaný Medzinárodný prognostický skórovací systém, revidovaný v roku 2012 (IPSS-R), ktorý rozdelil pacientov do 5 rizikových kategórií. Najvýznamnejším rizikovým parametrom v dĺžke prežívania a prechodu do akútnej leukémie je počet blastov v kostnej dreni (Tabuľka č. 2) (29).

Liečebná stratégia u pacientov s nízkym a stredným-1 rizikom
Hemopoetické rastové faktory
Erytropoézu stimulujúce látky (ESA) sa používajú u pacientov s nízkym alebo stredným-1 rizikom a s anémiou za účelom zvýšenia hladiny hemoglobínu a zníženia závislosti od transfúznej liečby. Avšak táto terapia stále ostáva off-label, pretože jej prínos nebol zatiaľ dokázaný v prospektívnej randomizovanej štúdii.
Lenalidomid
Lenalidomid sa používa na liečbu anemických pacientov s MDS s izolovanou del(5q). V štúdii MDS-003 bola hodnotená úloha lenalidomidu u nízkorizikových MDS pacientov s del(5q), ktorí boli závislí od transfúzie erytrocytov. U 76 % pacientov došlo poklesu závislosti od transfúzie a dvaja pacienti z troch sa stali nezávislými od transfúznej liečby. Navyše odpoveď na liečbu lenalidomidom sa dostavila skoro (medián 4,6 týždňov) a kompletná cytogenetická odpoveď je zdokumentovaná u 50 % pacientov (30).
Azanukleotidy
Liečba 5-azacitidínom a decitabínom nie je povolená v krajinách Európskej únie u pacientov s nízkym rizikom MDS. Štúdie, ktoré boli doteraz vykonané, nepreukázali prínos tejto terapie v nízkorizikovej skupine pacientov s MDS.
Imunosupresívna liečba
Existuje stále viac dôkazov, že lymfocyty a monocyty sa podieľajú na patogenéze MDS (31, 32). Približne 10 % pacientov s MDS má aj autoimunitné prejavy ochorenia. Patria sem laboratórne nálezy (hyper- alebo hypogamaglobulinémia, nález antinukleárnych protilátok) alebo klinické prejavy (napríklad vaskulitída). Niektoré štúdie in vitro preukázali, že T-lymfocyty a NK bunky od pacientov s MDS inhibujú rast autológnych progenitorových buniek. Okrem toho, klonálnu proliferáciu T-lymfocytov môžeme nájsť aj u pacientov s MDS. Navyše existuje aj značné prekrývanie medzi MDS a aplastickou anémiou. Niektoré štúdie ukázali, že približne jedna tretina pacientov s MDS reaguje na liečbu antitymocytovým globulínom (ATG) a/alebo cyklosporínom (CsA) (33, 34). Hoci sú publikované štúdie o účinnosti imunosupresívnej liečby s relatívne malým počtom pacientov, investigátori sa snažili identifikovať nezávislé parametre odpovede na liečbu. Ako nezávislý parameter sa ukázal vek pod 60 rokov a nízky počet trombocytov (33, 34).
Transfúzna a chelatačná liečba
Väčšina pacientov s MDS má anémiu a vyžaduje transfúznu liečbu. Čas nástupu potreby transfúznej liečby je významným prognostickým faktorom (35). Cieľom transfúznej liečby je zlepšenie kvality života, zabránenie vzniku príznakov anémie a ischemického poškodenia orgánov. Indikácia transfúzie sa neriadi konkrétnou hladinou hemoglobínu, ale vždy klinickým stavom pacienta, jeho vekom a prítomnými komorbiditami.
Keďže každá transfúzna jednotka erytrocytov obsahuje približne 250 mg železa, k rozvoju syndrómu z preťaženia železom môže dôjsť už po podaní 20 – 25 transfúznych jednotiek. Nadmerné hromadenie železa v organizme má negatívne následky a sprievodné komplikácie, medzi ktoré patria poškodenie pečene, srdca a žliaz s vnútornou sekréciou. Retrospektívne údaje ukázali, že chronická anémia spolu so syndrómom z preťaženia organizmu železom sa spája s kardiálnou dysfunkciou. Preto sú veľmi žiaduce stratégie v liečbe pacientov s MDS, ktoré zlepšujú anémiu a udržiavajú normálnu hladinu železa (36). Použitie chelatačnej liečby je preto jednou z nevyhnutných alternatív. Chelatačná liečba má začať u pacientov s MDS pri hladine feritínu od 1 000 – 2 500 µg/l. NCCN (Evidence-Based Cancer Guidelines) odporúčania hovoria o potrebe zvážiť možnosť chelatačnej liečby u pacientov s MDS s nízkym rizikom podľa IPSS-R alebo u pacientov, u ktorých sa predpokladá alebo už dostali viac než 20 transfúznych erytrocytových jednotiek a u ktorých je hladina feritínu vyššia než 2 500 µg/l. Cieľom je znížiť hladinu feritínu pod 1 000 µg/l (37, 38).
Liečebná stratégia u pacientov so stredným-2 a vysokým rizikom
Hypometylačná liečba
Vypnutie tumor supresorových a iných génov, ktoré riadia bunkový cyklus hypometyláciou, zohráva dôležitú úlohu v patogenéze a v progresii MDS (39, 40). Prvou študovanou látkou v polovici 90. rokov bol decitabín. Druhým študovaným liečivom bol azacitidín. Klinické výsledky s využívaním azacitidínu sú založené hlavne na dvoch multicentrických randomizovaných štúdiách vo fáze III. Ide o štúdie CALGB 9221 (41) a AZA-001 (42). V CALGB 9221 štúdii bol azacitidín (podávaný 75mg/m2/deň počas 7 po sebe nasledujúcich dňoch, každých 28 dní) porovnaný s najlepšou podpornou liečbou. Štúdie sa zúčastnilo 191 pacientov s MDS (klasifikovaných podľa FAB), z toho 46 % pacientov malo podľa IPSS riziko stredné-2 alebo vysoké. Priemerný vek pacientov v štúdii bol 68 rokov. Odpoveď na liečbu bola pozorovaná u 60 % pacientov liečených azacitidínom v porovnaní s 5 % pacientov, ktorí dostávali najlepšiu podpornú starostlivosť. Priemerná doba trvania odpovede bola 15 mesiacov a doba transformácie do AML bola dlhšia u pacientov liečených azacitidínom. V AZA-001 štúdii boli porovnávané tri tradičné režimy starostlivosti (CCR: intenzívna chemoterapia ako pri AML, terapia nízkymi dávkami cytozínarabinozidom a najlepšia podporná liečba) vs terapia azacitidínom. Do štúdie bolo zaradených 358 pacientov s MDS. Podľa IPSS tvorili pacienti so stredným-2 a vysokým rizikom 87 % prípadov MDS. Priemerný vek pacientov bol 69 rokov. Terapia azacitidínom signifikantne predĺžila medián prežívania (azacitidín: 24,5 mesiaca; CCR: 15,0 mesiacov, p = 0,0001). Progresia do AML bola oddialená. Taktiež sa znížili požiadavky na erytrocytárne koncentráty (p < 0,0001) a miera výskytu infekcií (p = 0,0032) bola signifikantne nižšia pri užívaní azacitidínu vs CCR. Výhody v prežívaní pacientov užívajúcich azacitidín (v porovnaním s pacientmi užívajúcimi nízku dávku cytozínarabinozidu alebo dostávajúcimi najlepšiu podpornú liečbu) boli pozorované bez ohľadu na vek pacientov (vrátane pacientov vo veku nad 75 rokov), na množstvo blastov v kostnej dreni (vrátane pacientov s 20 – 30 % blastov v kostnej dreni) či bez ohľadu na karyotyp.
Intenzívna chemoterapia
MDS je zvyčajne liečená protokolmi, ktoré sú podobné protokolom používaným pri terapii AML. Základom je vždy kombinácia cytozínarabinozidu s antracyklínom. V porovnaní s de novo AML sú kompletné remisie menej časté (40 až 60 %), trvajú kratšie (medián 10 až 12 mesiacov) a komplikácie vzniknuté počas liečby sú časté.
Alogénna transplantácia kmeňovými bunkami
Alogénna transplantácia kmeňovými bunkami je k dnešnému dňu jediným kuratívnym spôsobom liečby. Aj keď kompletná remisia môže byť dosiahnutá intenzívnou chemoterapiou, dlhodobý výsledok je bez následnej transplantácie zlý. Preto by pri stanovení diagnózy MDS malo byť našou prvou otázkou, či je pacient schopný podstúpiť transplantáciu hemopoetickými kmeňovými bunkami. U pacientov so stredným-2 a vysokým rizikom MDS je potrebná skorá transplantácia pre dosiahnutie čo najdlhšej doby prežívania (43). Najdôležitejším prognostickým faktorom výsledku transplantácie je cytogenetický profil pacienta. V nedávnej štúdii sa ukázalo, že prítomnosť komplexného karyotypu a/alebo monozomálneho karyotypu je predpokladom pre kratšie prežívanie po alogénnej transplantácii (44).
Záver
MDS tvorí skupina heterogénnych hematologických ochorení. Posudzovanie prognózy je individuálnym postupom. Rozhodnutie o vhodnej liečebnej stratégii je umením, najmä kvôli dostupnosti mnohých liečebných smerov. Napriek novým terapeutickým postupom a liečivám má dôležitú úlohu aj podporná starostlivosť (podávanie hemopoetických rastových faktorov, transfúzna, chelatačná terapia a antimykotická profylaxia).
Literatúra
- Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med. 2009; 361: 1872 – 85
- Schumacher HR, Nand S. Myelodysplastic Syndromes: Approach to Diagnosis and Treatment. New York: Igaku-Shoin Medical Publishers; 1995.
- Ma X, Does M, Raza A, Mayne ST. Myelodysplastic syndromes: incidence and survival in the United States. Cancer. 2007; 109: 1536 – 1542
- Rollison DE, Howlader N, Smith MT et al. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001–2004, using data from the NAACCR and SEER programs. Blood. 2008; 112: 45 – 52
- Goldberg SL, Chen E, Corral M et al. Incidence and clinical complications of myelodysplastic syndromes among United States Medicare beneficiaries. J Clin Oncol. 2010; 28: 2847 – 2852
- Cogle CR, Craig BM, Rollison DE, List AF. Incidence of the myelodysplastic syndromes using a novel claims-based algorithm: high number of uncaptured cases by cancer registries. Blood. 2011; 117: 7121 – 7125
- Deschler B, Lübbert M. Acute myeloid leukemia: epidemiology and etiology. Cancer. 2006; 107: 2099 – 2107
- Strom SS, Vélez-Bravo V, Estey EH. Epidemiology of myelodysplastic syndromes. Semin Hematol. 2008; 45: 8 – 13
- Paustenbach DJ, Bass RD, Price P. Benzene toxicity and risk assessment, 1972–1992: implications for future regulation. Environ Health Perspect. 1993; 101(suppl 6): 177 – 200
- Björk J, Albin M, Mauritzson N, Strömberg U, Johansson B, Hagmar L. Smoking and myelodysplastic syndromes. Epidemiology. 2000; 11: 285 – 291
- Nisse C, Haguenoer JM, Grandbastien B et al. Occupational and environmental risk factors of the myelodysplastic syndromes in the North of France. Br J Haematol. 2001; 112: 927 – 935
- Strom SS, Gu Y, Gruschkus SK, Pierce SA, Estey EH. Risk factors of myelodysplastic syndromes: a case-control study. Leukemia. 2005; 19: 1912 – 1918
- West RR, Stafford DA, Farrow A, Jacobs A. Occupational and environmental exposures and myelodysplasia: a case-control study. Leuk Res. 1995; 19: 127 – 139
- Rigolin GM, Cuneo A, Roberti MG et al. Exposure to myelotoxic agents and myelodysplasia: case-control study and correlation with clinicobiological findings. Br J Haematol. 1998; 103: 189 – 197
- Dalamaga M, Petridou E, Cook FE, Trichopoulos D. Risk factors for myelodysplastic syndromes: a case-control study in Greece. Cancer Causes Control. 2002; 13: 603 – 608
- Miller KB. Myelodysplastic syndromes. In: Wiernik PH, Goldman JM, Dutcher JP, Kyle RA, editors. Neoplastic Diseases of the Blood. 4. Cambridge: Cambridge University Press; 2003
- Ma X, Lim U, Park Y et al. Obesity, lifestylefactors, and risk of myelodysplastic syndromes in a large US cohort. Am J Epidemiol. 2009; 169: 1492 – 1499
- Ido M, Nagata C, Kawakami N et al. A case-control study of myelodysplastic syndromes among Japanese men and women. Leuk Res. 1996; 20: 727 – 731
- Yoshida K, Sanada M, Shiraishi Y et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011; 478: 64 – 9
- Nilsson L, Astrand-Grundstrom I, Arvidsson I, Jacobsson B, Hellstrom-Lindberg E, Hast R, Jacobsen SE. Isolation and characterization of hematopoietic progenitor/stem cells in 5q- deleted myelodysplastic syndromes: evidence for involvement at the hematopoietic stem cell level. Blood. 2000; 96: 2012 – 21
- Woll PS, Kjallquist U, Chowdhury O et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell 2014; 25: 794 – 808
- Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, Ebert BL Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011; 364: 2496 – 506
- Schanz J, Tuchler H, Sole F et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012; 30: 820 – 9
- Van den Berghe H, Cassiman JJ, David G, Fryns JP, Michaux JL, Sokal G. Distinct haematological disorder with deletion of long arm of no. 5 chromosome. Nature. 1974; 251: 437 – 8
- Boultwood J, Perry J, Pellagatti A et al. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia. 2010; 24: 1062 – 5
- Nikoloski G, Langemeijer SM, Kuiper RP et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010; 42: 665 – 7
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127(20): 2391 – 405
- Vondráková J. Myelodysplastický syndrome, diagnostika a léčba. Interní medicína pro praxi. 2010; 12(11): 535 – 539
- Greenberg PL, Attar E, Bennett JM. Myelodysplastic syndromes. et al. J Natl Compr Canc Netw. 2011; 9: 30 – 56
- List A, Dewald G, Bennett J, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006; 355: 1456 – 65
- Meers S, Kasran A, Boon L, Lemmens J, Ravoet C, Boogaerts M, Verhoef G, Verfaillie C, Delforge M. Monocytes are activated in patients with myelodysplastic syndromes and can contribute to bone marrow failure through CD40-CD40L interactions with T helper cells. Leukemia. 2007; 21: 2411 – 9
- Meers S, Vandenberghe P, Boogaerts M, Verhoef G, Delforge M. The clinical significance of activated lymphocytes in patients with myelodysplastic syndromes: a single centre study of 131 patients. Leuk Res. 2008; 32: 1026 – 35
- Broliden PA, Dahl IM, Hast R et al. Antithymocyte globulin and cyclosporine A as combination therapy for low-risk non-sideroblastic myelodysplastic syndromes. Haematologica. 2006; 91: 667 – 70
- Saunthararajah Y, Nakamura R, Nam JM, Robyn J, Loberiza F, Maciejewski JP, Simonis T, Molldrem J, Young NS, Barrett AJ. HLA-DR15 (DR2) is overrepresented in myelodysplastic syndrome and aplastic anemia and predicts a response to immunosuppression in myelodysplastic syndrome. Blood. 2002; 100: 1570 – 4
- Malcovati L, Della Porta MG, Pascutto C et al. Prognostic factors and life expectancy in myelodysplastic syndromes classified according to WHO criteria: a basis for clinical decision making. J Clin Oncol. 2005; 23: 7594 – 603
- Malcovati L, Della Porta MG, Pascutto C et al., prognostic Factors and Life Expectancy in Myelodysplastic Syndromes Classified According to WHO Criteria: A Basis for Clinical Decision Making. J Cof Clinical Oncology. 2005; 23(30): 7694 – 7703
- Greenberg PL, Attar E, Bennett JM et al. NCCN Guidelines: Myelodysplastic Syndromes. National Comperhensive Cancer Network. 2014; 2:MS1 – MS48
- Chudej J, Sokol J, Mikušková E et al. Manažment anémie u pacientov s myelodysplastickým syndrómom. Via Pract. 2014; 11(3-4): 108 – 111
- Quesnel B, Guillerm G, Vereecque R, Wattel E, Preudhomme C, Bauters F et al. Methylation of the p15(INK4b) gene in myelodysplastic syndromes is frequent and acquired during disease progression. Blood. 1998; 91(8): 2985 – 90
- Jiang Y, Dunbar A, Gondek LP, Mohan S, Rataul M, O’Keefe C et al. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood. 2009; 113(6): 1315 – 25
- Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002; 20(10): 2429 – 40
- Fenaux P, Mufti GJ, Hellström-Lindberg E, Santini V, Finelli C, Giagounidis A et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009; 10(3): 223 – 32
- Cutler CS, Lee SJ, Greenberg P et al. A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood. 2004; 104: 579 – 85
- an Gelder M, de Wreede LC, Schetelig J, van Biezen A, Volin L, Maertens J, Robin M, Petersen E, de Witte T, Kro- ger N. Monosomal karyotype predicts poor survival after allogeneic stem cell transplantation in chromosome 7 abnormal myelodysplastic syndrome and secondary acute myeloid leukemia. Leukemia. 2013; 27: 879 – 88

Tento článok sa nachádza v čísle invitro 04/2016
Hematológia
Tesne pred Vianocami vám prinášame jeden predčasný darček v podobe trinásteho čísla časopisu inVitro, ktoré je tentokrát venované hematológii. Aj v tomto čísle nájdete množstvo praktických odborných…





