


Epidermolysis bullosa hereditaria je primárne kožná choroba. U závažnejších subtypov sa stáva chorobou s multiorgánovým poškodením (vrátane uropoetického systému) v dôsledku infekcií, porúch výživy a sekundárneho postihnutia vnútorných orgánov.
Úvod
Choroba motýlích krídiel azda najviac zviditeľňuje všetky aspekty zriedkavých chorôb. Má nízku prevalenciu v populácii, teda spĺňa kritérium na zriedkavé choroby. Prevalencia je menej než 1 na 2 000 osôb. Choroba je charakterizovaná tvorbou pľuzgierov, ale s výraznou variabilitou prejavov a závažnosti. Poškodenie kože je niekedy vyslovene benígne a na opačnej strane spektra až ohrozuje život človeka. Rozlišujeme štyri základné typy a množstvo podtypov epidermolysis bullosa (EB) a napriek relatívne jednoduchej vizuálnej diagnostike sú presná diagnostika a klasifikácia zložité. EB je dedičná choroba, podmienená mutáciami v rôznych génoch. Dokumentované sú typy dominantnej a recesívnej dedičnosti. Aj liečba je veľmi variabilná, od jednoduchej prevencie tvorby pľuzgierov, napríklad nosením správnej obuvi, až po extrémne náročné ošetrovanie pacienta špeciálnymi prostriedkami. EB patrí ku chorobám, na ktoré sa výrazne poukazovalo v začiatkoch vývoja európskych iniciatív na zlepšenie diagnostiky a liečby zriedkavých chorôb (ZCH) ako na modelové príklady komplexnosti problematiky.
Najkomplexnejšia internetová stránka pre zriedkavé choroby s osobitnou klasifikáciou, ORPHANET, v súčasnosti uvádza 44 rozlíšiteľných EB chorôb a EB syndrómov (1). Najnovšia iniciatíva v oblasti ZCH je zameraná veľmi prakticky na európsku podporu diagnostiky a liečby prostredníctvom európskych referenčných sietí (ERN), pričom ERN Skin je jednou z 24 sietí uznaných a podporovaných Európskou komisiou (2). Základom ERN je zdieľanie skúseností, praktická pomoc v diagnostike a v manažovaní pacientov, ale aj výskum chorôb a podpora vývoja liekov na zriedkavé choroby. Vytvárajú sa zdieľané registre pacientov, vďaka ktorým sa ďalej rozvíjajú skúsenosti a vzdelávanie špecialistov. V Európe sa venuje tejto problematike okolo 80 pracovísk, a sú uvádzané 4 ERN pre túto chorobu (8). DEBRA (The Dystrophic Epidermolysis Bullosa Research Association of America – DEBRA of America) je celosvetovo rozšírená pacientska organizácia zameraná na všestrannú podporu problematiky EB vrátane výskumu a podpory rodín, ktorá pôsobí aj na Slovensku (3, 4).
Dedičné formy EB tvoria širšiu skupinu chorôb. Všetky typy a podtypy EB sú zriedkavé, incidencia sa v USA uvádza 1 na 53 000 novorodencov, prevalencia v populácii je 1 na 125 000. Obdobné epidemiologické štatistiky sú aj z krajín Európy.
Etiológia a klasifikácia
Variabilita prejavov je od lokálnej tvorby pľuzgierov na rukách a nohách až po generalizované postihnutie kože a dutiny ústnej a poškodenie viacerých vnútorných orgánov. Podmieňuje to štruktúrová fragilita epidermis a niektorých ďalších tkanív. Štyri hlavné typy sú EB simplex (EBS), junkčná EB (JEB), dystrofická EB (DEB) a Kindlerovej syndróm (Tabuľka č. 1). Klasifikácia EB u pacienta do niektorého z množstva podtypov je však komplexná. Použije sa opis podrobného klinického nálezu s lokalizáciou a charakteristikou kožných zmien, s vývojom a rozsahom kožných zmien, s vekom nástupu príznakov a s nálezmi o celkovom zdravotnom stave. V rodinnej anamnéze pátrame po výskyte aj minimálnych príznakov u členov rodiny s cieľom vyhodnotiť pravdepodobný typ dedičnosti subtypu. Imunohistochémia je zameraná na antigénnu štruktúru a na farbenie štruktúr a je to podstatná laboratórna metóda klasifikácie subtypov. U jednotlivých subtypov dnes poznáme príslušné poškodené proteíny. Elektrónová mikroskopia prispieva k diagnostike subtypov identifikáciou vrstvy epidermis, v ktorej sa pľuzgiere tvoria. Každý subtyp má známe mutácie v príslušnom géne kódujúcom niektorý proteín. Tieto proteíny sú osobitne zapojené do udržiavania štrukturálnej stability keratinocytov a adhézie keratinocytov na dermis. Na základe genotypovej a fenotypovej variability je opísaných viac než 30 odlišných typov.

Konsenzuálna klasifikácia EB z roku 2008 podrobne opisuje každý subtyp (5). Odvtedy však bolo zistených niekoľko ďalších génov pre EB a ďalších subtypov pre EB a navrhuje sa inovovaná klasifikácia s väčším dôrazom na laboratórne vyšetrenia proteínov a génov (Tabuľka č. 2) (6).
Ilustratívny pohľad na základné poznatky použité na triedenie hlavných typov a podtypov poskytuje tabuľka, v ktorej sa ku nomenklatúre z roku 2008 pridávajú poznatky z posledných rokov výskumu (Tabuľka č. 3) (6). V Tabuľke č. 3 sú použité poznatky o mutáciách v génoch etiologicky asociovaných s konkrétnymi subtypmi EB.
Klinický obraz
Okrem mechanicky fragilnej kože a ľahkej tvorby pľuzgierov sa vyskytujú ďalšie príznaky: milia (jemné biele papuly pripomínajúce cysty alebo pustulky), dystrofia alebo chýbanie nechtov, zvyčajne atrofické jazvy, niekedy granulačné tkanivo v mechanicky namáhanej koži, keratoderma rúk a nôh a dyspigmentácie. Zriedkavo sa vyskytnú chýbanie alebo zníženie množstva vlasov, hypohydróza alebo hyperhydróza (7). Choroba epidermolysis bullosa bola od začiatkov opisov tejto choroby známa veľkou variabilitou. Výskumy o zmenách ultraštruktúry a o poškodených proteínoch metódami elektrónovej mikroskopie a histochémie spresnili charakteristiky subtypov a vyčlenili nové subtypy. Podobné spresnenie typológie prináša aj výskum génov zameraný na postihnuté proteíny.
Intraepiteliálna tvorba pľuzgierov je základným znakom EBS – epidermolysis simplex. EBS je ďalej subklasifikovaná na subtyp suprabazálny (patria sem subtypy: EB simplex superficialis – instabilita je subkorneálne, EB lethal acantholytic – štiepenie na úrovni suprabazálnej, EBS pri plakofilínovej deficiencii – štiepenie je uprostred epidermis) a subtyp EBS bazálny (patria sem lokalizovaná EBS, EBS Dowling-Meara, generalizovaná EBS, autozómovo recesívna EBS – všetky majú štiepenie na úrovni bazálnych keratinocytov). U niektorých subtypov EBS nachádzame charakteristické ultraštruktúrové znaky v elektrónovej mikroskopii, typické znaky pri detekcii antigénov, známe sú poškodenia cieľových proteínov a podľa inovovaného návrhu klasifikácie z roku 2014 aj mutácie v príslušných génoch. V skupine suprabazálnych EBS podtypov sú poškodené proteíny: transglutamináza 5, dezmoplakín, plakoglobín a plakofilín. V skupine bazálnych EBS podtypov boli zistené poškodenia proteínov: keratín 5, keratín 14, exophollin 5, plektín, bullous pemphigoid antigen-1. Mutácie pre túto skupinu boli zistené v 9 génoch. Na základe rozširovania poznatkov o poškodených cieľových proteínoch a o mutáciách v príslušných génoch sa navrhuje aj podrobnejšia subklasifikácia, napríklad do EBS sa sem zaraďuje 18 subtypov (6).

Dystrofická EB sa člení na podtypy podľa typu dedičnosti, dominantne dedičná forma a recesívne dedičná forma. Dominantne dedičná forma (DDEB) má formu DDEB generalizovanú a DDEB – bulóznu dermatolýzu novorodencov. Recesívne dedičná forma dystrofickej EB má formu RDEB ťažká generalizovaná a RDBE generalizovaná stredne ťažká. Vo všetkých štyroch subtypoch je instabilita v sublaminárnej vrstve. Obraz v ultraštruktúre podľa elektrónovej mikroskopie je u jednotlivých podtypov rôzny. Mutácie sú zistené v géne pre kolagén VII, COL7A1. Poškodený je proteín kolagén VII, na rôznom stupni. Opisuje sa normálne alebo redukované farbenie, granulárne farbenie v bazálnych keratinocytoch, niekedy chýba farbenie pozdĺž dermoepidermálneho spojenia. U DDEB a RDEB bulóznej dermatolýzy novorodencov je nález variabilný aj podľa aktivity choroby. Na základe morfológie dystrofických zmien a klinických opisov je v navrhovanej novej klasifikácii vygenerovaných 14 subtypov (6).
Kindlerovej syndróm je charakterizovaný štiepením vo viacerých vrstvách epidermis vo vzorke kože. Mutácie sú zistené v géne FERMT1 (KIND1) kódujúcom proteín kindlín 1.
Diagnostika

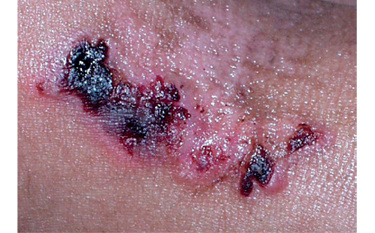
Liečba a prevencia
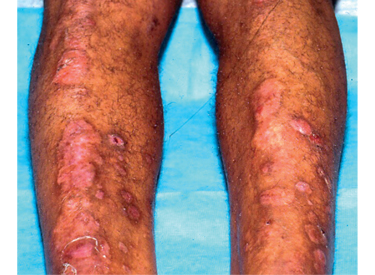

Záver
Choroba epidermolysis bullosa má širokú klinickú variabilitu. Pre lekára je veľmi dôležité zaradenie subtypu EB do najnovšej klasifikácie. Klasifikácia nie je samoúčelná, v mnohom prognózuje priebeh choroby a života pacienta. Presnejšiu prognózu možno získať len správnym zaradením do subtypu EB. Zaradenie do správneho subtypu sa odvíja od použitia histochemického farbenia proteínov, elektrónovej mikroskopie a molekulárnej genetiky. Prebieha intenzívny výskum génovej terapie. Pri poznaní typu dedičnosti a príslušnej mutácie u pacienta a v rodine je možná prenatálna genetická diagnostika a to aj v predimplantačnom období prostredníctvom asistovanej reprodukcie. Počas života je to náročná, až extrémne náročná celoživotná starostlivosť o pacienta. Najťažšie typy si v medicíne vyslúžili nelichotivé označenie najstrašnejšej choroby, akú ľudstvo pozná.
Literatúra
- ORPHANET – dostupné na: http://www.orpha.net/consor/cgi-bin/index.php
- European reference networks. Electronic version: ISBN 978-92-79-65502-9
- Epidermolysis bullosa – dostupné na: www.debra.org
- Debra Slovakia – dostupné na: http://www.debra-slovakia.org/
- Fine JD, Eady RAJ, Bauer JA, et al.: The classification of inherited epidermolysis bullosa (EB): report of the Third international Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol 2008, 58:931-950
- Fine JD, et al. : Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification, J AmAcad Dermatol june 2014,1103-1126 http://dx.doi.org/10.1016/j.jaad.2014.01.903
- Jo-David Fine: Inherited epidermolysis bullosa: Orphanet Journal of Rare Diseases 2010, 5:12, doi: 10.1186/1750-1172-5-12
- Chovancová D., Kaiserová Lˇ.: Epidermolysis bullosa (choroba motýlich krídiel) Pediatr. prax, 2007; 2: 102–104. dostupné na: www.solen.eu

