
Idiopatická pľúcna fibróza je špecifickou formou chronického fibrotizujúceho intersticiálneho pľúcneho procesu, ktorá patrí k najzávažnejším pľúcnym ochoreniam, najmä kvôli veľmi zlej prognóze a refraktérnosti na doteraz známu liečbu. V posledných rokoch došlo k významným zmenám v klasifikácii, diagnostike a v liečbe tejto závažnej choroby. Nové antifibrotické lieky dávajú nádej na zlepšenie kvality života, ako aj na predĺženie života pacientov s touto zákernou chorobou.
Úvod
Intersticiálne pľúcne procesy (IPP) tvoria početnú heterogénnu skupinu ochorení, ktorých spoločným prejavom je difúzne postihnutie pľúcneho tkaniva. Medzi IPP sa zaraďuje viac než 150 nozologických jednotiek, ktoré majú často podobný klinický, rádiologický a funkčný obraz. U mnohých IPP je príčina známa (inhalácia organických a anorganických látok, radiačné žiarenie, lieky, neoplázie, infekčné agensy), ale asi 40 – 50 % IPP vzniká z nejasných príčin (systémové ochorenia ako sarkoidóza, histiocytóza X, systémové choroby spojiva, vaskulitídy, idiopatické intersticiálne pneumónie).
Idiopatické intersticiálne pneumónie (IIP) predstavujú pomerne heterogénnu podskupinu intersticiálnych pľúcnych procesov neznámej etiológie, ktoré sú charakterizované rôznym stupňom zápalového procesu v alveolách (alveolitída) so súčasne prebiehajúcou fibrotickou prestavbou pľúcneho interstícia (fibróza) a práve tento pomer v konečnom dôsledku určuje prognózu daného ochorenia. Medzi IIP sa zaraďuje aj idiopatická pľúcna fibróza (IPF), ktorá patrí k najzávažnejším pľúcnym ochoreniam (Tabuľka č. 1).

Definícia
IPF je definovaná ako špecifická forma chronickej progredujúcej fibrotizujúcej intersticiálnej pneumónie neznámej etiológie vyskytujúcej sa u dospelých jedincov, ktorá postihuje iba pľúca a je spojená s histopatologickým a/alebo rádiologickým obrazom obvyklej intersticiálnej pneumónie (usual interstitial pneumonia, UIP) (2, 3).
Incidencia a prevalencia
Údaje o incidencii a prevalencii IPF sa celosvetovo výrazne líšia. Predpokladá sa, že postihuje asi 5 miliónov ľudí na celom svete. Podľa epidemiologických štúdií sa incidencia uvádza medzi 6,8 a 16,3/100 000 obyvateľov a prevalencia 2 – 29/100 000 obyvateľov (1). V Európe sa incidencia uvádza medzi 0,22 a 7,56/100 000 a prevalencia 1,25 až 25,6/100 000 obyvateľov. Predpokladá sa, že v súčasnosti žije v Európe 80 000 až 111 000 ľudí s IPF a každý rok sa v EÚ diagnostikuje približne 30 000 až 35 000 nových prípadov. Zvýšená prevalencia sa pozoruje v posledných rokoch pravdepodobne kvôli optimalizácii diagnostických metód a kvôli nárastu očakávanej dĺžky života (2, 3, 5, 6).
Mortalita IPF je vysoká, v USA dosahuje v mužskej populácii 61,2 úmrtí na 1 000 000 obyvateľov a u žien 54,5 na 1 000 000 obyvateľov. Príčinou úmrtí je najčastejšie progresia IPF (60 %), ďalšie príčiny sú ochorenia koronárnych tepien, pľúcna embólia a rakovina pľúc.
Podľa aktuálnych údajov, incidencia a prevalencia IPF nezávisia od geografických, sociálnych ani rasových faktorov. Incidencia IPF sa zvyšuje s vekom, väčšina pacientov je nad 50 rokov a postihuje viac mužov než ženy. Obvykle sa vyskytuje sporadicky, familiárne prípady sú vzácne, nepresahujú 5 % všetkých prípadov.
Etiológia
Etiológia IPF nie je známa, ale je pravdepodobne spôsobená účinkom rôznych faktorov u geneticky predisponovaných subjektov. Ide o odpoveď pľúcneho tkaniva na rôzne stimuly, ktoré spôsobujú poškodenie pľúcneho interstícia a vedú k progredujúcej a ireverzibilnej fibróze. Existuje rad faktorov, ktoré majú vplyv na vznik a rozvoj IPF. Medzi potenciálne rizikové faktory patria cigaretový dym, expozícia prachu s obsahom niektorých minerálov a kovov (mosadz, oceľ, olovo, silikón), drevín (borovica, breza), prekonaná vírusová infekcia (EBV, CMV, vírus chrípky, parachrípky, hepatitídy C, HIV), chronický gastroezofageálny reflux s mikroaspiráciou a genetická predispozícia. Klinicky významnými genetickými zmenami sú mutácie v génoch pre telomerázy (TERT, TERC), ktoré sú bežnejšie vo familiárnych formách, mutácie v géne pre surfaktantový proteín C a v promótorovej oblasti 5B mucínu (MUC5B) (7).
Diagnostika a klinický obraz
Konečná diagnóza IPF vyžaduje:
a) vylúčenie všetkých iných foriem intersticiálnych pneumónií, ktoré môžu mať veľmi podobný rádiologický, klinický a histopatologický obraz – predovšetkým iné idiopatické intersticiálne pneumónie, systémové ochorenia spojiva a intersticiálne pľúcne procesy spojené s expozíciou vplyvom prostredia,
b) histopatologický a/alebo rádiologický obraz obvyklej intersticiálnej pneumónie (UIP).
V súčasnosti je všeobecne akceptovaným odporúčaním na stanovenie diagnózy IPF multidisciplinárne hodnotenie, na ktorom by sa mali zúčastňovať pneumológovia, rádiológovia a patológovia so skúsenosťami v diagnostike a v liečbe IPP. Na základe len rádiologického alebo patologického nálezu UIP, bez poznania klinických a laboratórnych výsledkov, nie je možné stanoviť diagnózu IPF.
Typický klinický obraz IPF je charakterizovaný pomalým nástupom s progredujúcou dýchavicou pri námahe sprevádzanou neproduktívnym kašľom. Čas od začiatku symptómov po stanovenie diagnózy je variabilný a pohybuje sa medzi 6 mesiacmi a dvoma rokmi. V pokročilejších štádiach sa objavuje hypoxémia a cyanóza. U niektorých pacientov sa vyskytnú epizódy akútnych exacerbácií IPF charakterizované náhlym výrazným zhoršením dušnosti. Prítomnosť systémových príznakov by mala viesť k podozreniu na alternatívnu diagnózu. Pri fyzikálnom vyšetrení sú typické krepitácie na bázach pľúc pri auskultácii asi u 90 % pacientov a paličkovité prsty sa zistia asi v 50 % prípadov. Výsledok kardiologického vyšetrenia môže byť normálny, alebo podľa pokročilosti ochorenia môže byť prítomná pľúcna hypertenzia, cor pulmonale chronicum a pravostranná srdcová dekompenzácia.
IPF má obvykle progredujúci priebeh. U časti pacientov má ochorenie veľmi rýchly priebeh, u časti môže prebiehať pomaly s postupným poklesom pľúcnych funkcií, v ktoromkoľvek štádiu ochorenia sa môžu vyskytnúť akútne exacerbácie s veľmi zlou prognózou. Medián prežívania od stanovenia diagnózy sa pohybuje medzi 2,5 a 3,5 rokmi.
Laboratórne vyšetrenia
Neexistujú žiadne špecifické laboratórne vyšetrenia, ktoré by potvrdili diagnózu IPF. U všetkých pacientov s podozrením na IPF treba vylúčiť systémové ochorenia spojiva, preto by sa aj pri absencii špecifických príznakov alebo symptómov ochorení spojivového tkaniva mala vykonať skríningová sérologická diagnostika na prítomnosť systémového ochorenia (reumatoidný faktor, antinukleárny faktor, protilátky proti citrulínovým peptidom – ACPA, SS-A, SS-B, PM/Scl-75, PM/Scl-100) a vyšetrenie protilátok proti organickým antigénom na vylúčenie exogénnej alergickej alveolitídy (EAA) (2, 8).
Funkčné vyšetrenie pľúc
U každého pacienta s podozrením na IPP by mali byť realizované funkčné vyšetrenia pľúc, a to minimálne spirometrické vyšetrenie, doplnené bodypletyzmografiou a vyšetrenie difúznej kapacity pľúc (transfer faktor – TLco). Typickými funkčnými poruchami u pacientov s IPF sú reštrikčná ventilačná porucha so znížením vitálnej kapacity a totálnej kapacity pľúc (TLC) a zníženie TLco a pľúcnej poddajnosti. Reziduálny objem (RV) je u pacientov s IPF obvykle zachovaný a pomer RV/TLC je často zvýšený. U malej časti pacientov môže byť prítomná aj obštrukčná ventilačná porucha – u podskupiny pacientov s IPF a zároveň s prítomným pľúcnym emfyzémom. Difúzna kapacita je často znížená už v počiatočných štádiách ochorenia, keď ešte pľúcne objemy nie sú redukované. Vyšetrenie krvných plynov v arteriálnej krvi môže byť v úvode ochorenia v medziach normy a k poklesu saturácie krvi kyslíkom a hypoxémii dochádza až pri záťaži. S progresiou ochorenia dochádza k hypoxémii aj v pokoji, keď už je u pacientov indikovaná dlhodobá kyslíková liečba.
Bronchoalveolárna laváž (BAL)
Podľa posledného konsenzu ATS/ERS, JRS/ALAT 2011 nie je rutinné vyšetrenie BAL odporúčané (2). Napriek tomu, v niektorých prípadoch môže byť vyšetrenie BAL veľmi užitočné pri diferenciálnej diagnostike IPP. V mnohých prípadoch môže potvrdiť alternatívnu diagnózu (alveolárna proteinóza, malignity, oportúnne infekcie, alveolárna hemorágia) a napomáha pri odlíšení IPF od chronickej hypersenzitívnej pneumonitídy (HP) alebo nešpecifickej intersticiálnej pneumónie (NSIP).
Pre IPF je typické zmnoženie neutrofilov, obvykle s malou prímesou eozinofilov, v tekutine získanej pri BAL. Naopak, zvýšenie lymfocytov skôr podporuje diagnózu HP alebo NSIP.
Pľúcna biopsia
V niektorých prípadoch treba k definitívnej diagnóze IPF pľúcnu biopsiu. Transbronchiálna biopsia obvykle nie je prínosná pre diagnostiku IPF kvôli malému množstvu získaného pľúcneho tkaniva. V posledných rokoch sa preferuje cielený odber materiálu pri vyšetrení pomocou endobronchiálneho ultrazvuku (EBUS) alebo transbronchiálna kryobiopsia, pomocou ktorej je možné získať väčšiu vzorku tkaniva a jej výťažnosť je mnohonásobne vyššia (8, 9). Najvýťažnejšia vzorka pľúcneho tkaniva sa môže získať chirurgickou biopsiou (obyvkle cestou videoasistovanej torakoskopie – VATS), avšak v súčasnosti sa odporúča iba pri diagnostických nejasnostiach, kvôli vyššiemu riziku morbidity a mortality a kvôli zvýšenému výskytu akútnych exacerbácií po tomto výkone (10). Chirurgickú pľúcnu biopsiu netreba vykonávať pri typickom obraze UIP pri HRCT vyšetrení pľúc a pri vylúčení iných príčin IPP.
Zobrazovacie metódy
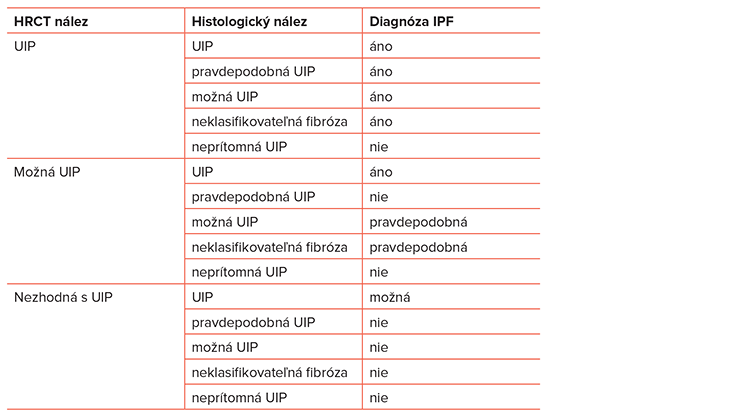
Skiagram hrudníka je prvým vyšetrením pri podozrení na IPP. Charakteristickými znakmi sú retikulácia a retikulonodulácia s maximom v dolných pľúcnych poliach a v pokročilejších štádiách aj zmenšenie objemu pľúc s vyšším postavením bránice. Pri každom podozrení na IPP treba CT vyšetrenie pľúc s vysokou rozlišovacou schopnosťou (HRCT). HRCT je základnou zložkou diagnostického procesu IPF. Typický obraz UIP na HRCT tvoria retikulárne abnormality a plástové pľúca s trakčnými bronchiektáziami alebo bez nich, vyskytujúce sa predominantne subpleurálne a bazálne. Opacity typu mliečneho skla sú väčšinou malého rozsahu alebo nie sú prítomné. Podľa zmien v HRCT obraze by mal rádiológ stanoviť, či nález zodpovedá obrazu UIP, možnému obrazu UIP, alebo je nález nekonzistentný s UIP (Tabuľka č. 2).

Pozitívna prediktívna hodnota HRCT pri diagnostike UIP je 90 – 100 %. Nález UIP nie je typický iba pre IPF. Môže byť prítomný aj pri chronickej hypersenzitívnej pneumonitíde, azbestóze a pri niektorých ochoreniach spojivového tkaniva. HRCT tiež umožňuje zhodnotiť prítomnosť asociovaných komorbidít (emfyzém, pľúcnu hypertenziu a rakovinu pľúc).
Histopatologický obraz
Ak HRCT nevykazuje definitívny obraz typický pre UIP, definitívna diagnóza by mala byť potvrdená histologickým vyšetrením pľúcneho tkaniva. Pre UIP sú typické deštrukcia architektúry pľúcneho parenchýmu, fibróza s roztrúsenými fibroblastickými ložiskami, heterogenita postihnutia pľúc s ložiskami nepostihnutého parenchýmu, striedajúcimi sa s ložiskami intersticiálneho zápalu, fibrózy a zmeny typu plástových pľúc. Preto aj dostatočne veľká vzorka tkaniva, avšak odobratá z nesprávneho miesta, nemusí prispieť k stanoveniu diagnózy.
V akcelerovanej fáze ochorenia je okrem histologického nálezu typického pre UIP prítomná aj prímes najrôznejších akútnych lézií (organizujúca sa pneumónia, difúzne alveolárne poškodenie, kapilaritída, zápal). Histologický nález by mal zhodnotiť patológ ako UIP, pravdepodobná UIP, možná UIP, neprítomná UIP alebo neklasifikovateľná fibróza.
Pri systémových ochoreniach spojiva (napríklad reumatoidná artritída a sklerodermia), chronickej hypersenzitívnej pneumonitíde, pri pneumonitídach vyvolaných liečivami či pri azbestóze je možné pozorovať histologický nález, ako aj HRCT obraz nerozoznateľný od UIP. Kvôli tomu sa UIP nemôže interpretovať priamo ako IPF bez toho, aby boli všetky tieto choroby vylúčené.
Podľa oficiálneho konsenzu ATS/ERS/JRS/ALAT z roku 2011 sa odporúča, aby diagnóza idiopatickej intersticiálnej pneumónie bola založená na konsenze medzi klinikom, rádiológom a patológom. Pri nezhode HRCT obrazu a histologického nálezu rozhoduje klinický nález a definitívnu diagnózu stanoví pneumológ. Ale aj tu sú možnosti diagnózy IPF interpretované ako „istá, pravdepodobná, možná a vylúčená“ (Tabuľka č. 3).
Liečba
Pred začiatkom liečby u pacientov s diagnózou IPF sa vždy musia hodnotiť prognostické faktory, štádium ochorenia a komorbidity. Terapeutické možnosti zahŕňajú farmakologickú liečbu, liečbu komplikácií a komorbidít, ktoré zhoršujú ochorenie (gastroezofageálny reflux, respiračné infekcie, pľúcna hypertenzia, fajčenie), rehabilitáciu, transplantáciu pľúc v prípadoch, ktoré spĺňajú kritériá a paliatívnu liečbu v záverečnej fáze ochorenia.
Farmakologická liečba
Do roku 2011 nebol dostupný žiadny liek, ktorý by mohol zmeniť priebeh ochorenia. Použitie glukokortikoidov a imunomodulátorov (azatioprín alebo cyklofosfamid) spolu s N-acetylcysteínom (NAC) bolo dlho odporúčané ako štandard liečby, aj napriek chýbajúcim dôkazom o ich efektivite. V roku 2012 bola v štúdii PANTHER potvrdená vyššia mortalita v skupine pacientov, ktorí boli lieční touto kombináciou, v porovnaní s vetvou pacientov liečených iba NAC alebo placebom. Kvôli tomu sa táto kombinácia pre pacientov s IPF už neodporúča.
Základom farmakologickej liečby IPF sa v roku 2011 stala antifibrotická liečba, od ktorej sa očakáva spomalenie fibrotizácie pľúcneho parenchýmu a s tým súvisiaci pokles pľúcnych funkcí a spomalenie priebehu ochorenia. V súčasnosti sú k dispozícii dve molekuly, ktoré môžu spomaliť fibrotizáciu a ktoré prvýkrát v histórii chorých s IPF dokázali v klinických štúdiách predĺžiť celkovú dobu prežívania – pirfenidon a nintedanib.
Pirfenidon
Pirfenidon (Esbriet) pravdepodobne inhibuje fibroproliferáciu ovplyvnením expresie transformujúceho rastového faktora beta. V klinických štúdiách sa potvrdila efektivita v zmysle zníženia rýchlosti poklesu pľúcnych funkcií a mortality u pacientov s IPF. Na základe výsledkov klinických štúdií sa v súčasnosti odporúča jeho použitie u pacientov s miernou až stredne ťažkou formou IPF, s FVC > 50 % a TLco > 35 % náležitých hodnôt v dávke 2403 mg/deň (3-krát 3 tablety a 267 mg denne).

Nintedanib
Nintedanib (Ofev) je silný inhibítor tyrozínkinázy, ktorý pôsobí na vaskulárny endotelový rastový faktor (VEGF), na receptory rastového faktora odvodené z krvných doštičiek (PDGF) a na fibroblastový rastový faktor (FGF). Výsledky klinických štúdií potvrdili zníženie poklesu pľúcnych funkcií, pokles počtu exacerbácií a zlepšenie kvality života pacientov s IPF. Podáva sa 2-krát denne 150 mg perorálne a je indikovaný u pacientov s FVC > 50 % a TLco > 30 % náležitých hodnôt. Obidva lieky sú registrované aj na Slovensku, ale nie sú zaradené do kategorizačného zoznamu, preto je ich predpísanie viazané na súhlas zdravotných poisťovní. Indikácia liekov je viazaná na centrá pre diagnostiku a liečbu IPF, ktoré sú na Slovensku na Klinike pneumológie, ftizeológie a funkčnej diagnostiky SZU a UN Bratislava, na Klinike pneumológie a ftizeológie UN Martin, na Klinike pneumológie a ftizeológie UN L. Pasteura Košice, v NÚTaRCh Vyšné Hágy a v Špecializovanej nemocnici sv. Svorada Zobor.
Liečba kašľa (najmä nočný kašeľ) a dýchavice sú rozhodujúcimi faktormi na udržanie prijateľnej kvality života u pacientov s IPF. Kodeín a nízke dávky morfínu, aj vo forme náplastí s pomalým uvoľňovaním, môžu zmierniť pocit dušnosti a pretrvávajúceho kašľa u pacientov s pokročilým ochorením.
U pacientov s gastroezofageálnym refluxom (GER) sa odporúča podávanie inhibítorov protónovej pumpy, keďže sa predpokladá, že GER v dôsledku mikroaspirácií môže mať úlohu v patogenéze IPF, ako aj podiel na progresii ochorenia.
Nefarmakologická liečba
Rehabilitácia
Rehabilitácia sa ukazuje ako účinný a bezpečný spôsob na zlepšenie výkonnosti, zmiernenie dušnosti a zlepšenie kvality života pacientov s IPF. Pozitívne účinky rehabilitácie na dlhodobé prežívanie pacientov s IPF nie sú zdokumentované. Ukazuje sa, že pacienti s IPF získajú dlhodobejší benefit z rehabilitačných programov, keď je ochorenie ešte mierne, preto sa odporúča zaradiť pacientov s IPF do rehabilitačných programov v začiatočných štádiách ochorenia.
Dlhodobá domáca oxygenoterapia (DDOT)
DDOT je indikovaná u pacientov v pokročilých štádiách ochorenia a s trvalou hypoxémiou. Indikácia DDOT u pacientov s IPF a s hypoxémiou v pokoji vychádza z extrapolácie záverov štúdií vykonaných u pacientov s chronickou obštrukčnou chorobou pľúc a s chronickým respiračným zlyhaním. Neexistujú presvedčivé údaje podporujúce použitie ambulantnej kyslíkovej terapie u pacientov, ktorí desaturujú len počas záťaže, bez hypoxémie v pokoji. Z dôvodu nedostatku špecifických údajov u pacientov s IPF sa odporúča DDOT, ak sa zistí významná hypoxémia v pokoji alebo pri záťaži (SaO2 ≤ 88 %) (11).
Transplantácia pľúc
Transplantácia pľúc je jedinou liečbou pokročilého IPF, ktorej výsledkom je významné funkčné zlepšenie a zlepšenie prežívania a ktorá stále presahuje výsledky najlepšej dostupnej farmakologickej liečby. Ročné prežívanie po transplantácii pľúc je okolo 80 % a päťročné prežívanie je viac než 50 %. Transplantácia pľúc by sa preto mala zvažovať u všetkých pacientov už v čase stanovenia diagnózy alebo minimálne u pacientov pri poklese pľúcnych funkcií pod kritické hodnoty (pri poklese totálnej pľúcnej kapacity a vitálnej kapacity pod 50 % náležitých hodnôt, TLco pod 30 % náležitých hodnot a PaO2 pod 6 kPa).
Záver
IPF je samostatná jednotka v rámci IIP so špecifickým manažmentom a stále nepriaznivou prognózou, ktorá je porovnateľná s malígnymi onkologickými ochoreniami. V posledných rokoch došlo k významným zmenám v klasifikácii, v diagnostike a v liečbe IPF. V súčasnosti existujú dva registrované lieky na liečbu IPF s dokázanou účinnosťou na spomalenie prirodzeného priebehu ochorenia, a to pirfenidón a nintedanib. Výskum v oblasti patofyziológie tejto závažnej choroby a vývoj nových liekov dávajú nádej na zlepšenie kvality života, ako aj na predĺženie života pacientov s touto zákernou chorobou.
Literatúra
- An official ATS/ERS statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 21 2013; 188: 733 – 748.
- Raghu, G., Collard, H. R., Egan, J. J., Martinez, F. J., Behr, J., Brown, K. K.: An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management Am. J. Respir. Crit. Care Med., 183 (2011), s. 788 – 824.
- Raghu, G. et al.: An official ATS/ERS/JRS/ALAT Clinical Practice guideline: Treatment of Idiopathic Pulmonary fibrosis: Executive Summary. An update of the 2011 Clinical Practice Guideline. Am J Resp Crit Care Med. 2015;192: 238 – 48.
- American Thoracic Society/European Respiratory Society. International multidisciplinary consensus classification of the idiopathic interstitial pneumonias, Am J Respir Crit Care Med 2002; 165: 277 – 304.
- Ley, B., Collard, H.: Epidemiology of Idiopathic Pulmonary Fibrosis, Clinical Epidemiology 2013, s. 483 – 492.
- Eurostat News Release. Available at ec.europa.eu/eurostat. Accessed on 18 August 2014.
- Mcneal, D., Schwartz, A.: The genetics and environmental causes of pulmonary fibrosis, Proc Am Thorac Soc, 9 (2012), s. 120 – 125.
- Vašáková, M., Polák, J., Matěj. R.: Intersticiální plicní procesy. 2.aktualizované a rozšířené vydaní. Maxdorf Jessenius (2016), 132 – 157.
- Babiak, A., Hetzel, J., Krishna, G., Fritz, P., Moeller, P., Balli, T.: Transbronchial cryobiopsy: a new tool for lung biopsies Respiration, 78 (2009), s. 203 – 208.
- Kondoh, Y., Taniguchi, H., Kitaichi, M., Yokoi, T., Johkoh, T., Oishi, T., Kimura, T., Nishiyama, O., Kato, K., du Bois, R. M.: Acute exacerbation of interstitial pneumonia following surgical lung biopsy. Respir Med 2006;100: 1753 – 1759.
- Visca, D., Montgomery, A., de Lauretis, A., Sestini, P., Soteriou, H., Maher, T. M.: Ambulatory oxygen in interstitial lung disease Eur. Respir. J., 38 (2011), s. 987 – 990.
