Waldenströmova makroglobulinémia (WM) je veľmi vzácne ochorenie s rôznorodým klinickým obrazom. V typických prípadoch postihuje najmä kostnú dreň, lymfatické uzliny a slezinu (7 – 30 %). Ochorenie je nevyliečiteľné, ale má pomerne priaznivú prognózu. Medián celkového prežitia je viac než 10 rokov.
Úvod
V roku 1944 Ján Gosta Waldenström, švédsky lekár, prvýkrát opísal prípad dvoch pacientov, ktorí mali opakované krvácania z nosa a úst, mali zväčšené lymfatické uzliny, trombocytopéniu, zrýchlenú sedimentáciu erytrocytov, zvýšenú viskozitu séra, zvýšený počet lymfoidných buniek v kostnej dreni a normálny nález na snímkach kostí (13). Tento popis sa stal základom pre rozpoznanie choroby, ktorú dnes nazývame Waldenströmova makroglobulinémia. Vzhľadom na malú incidenciu WM zostala dlho definovaná len ako choroba s prítomnosťou zvýšenej koncentrácie monoklonového imunoglobulínu typu IgM (M‑IgM) (3).
Až WHO klasifikácia v roku 2008 definovala histopatologický základ tejto choroby – lymfoplazmocytový lymfóm infiltrujúci kostnú dreň, ktorý produkuje M‑IgM. Okrem WHO sa tejto chorobe venuje najmä medzinárodná pracovná skupina – International Workshop on Waldenström Macroglobulinemia (IWWM), ale aj ďalší (3, 7).
Epidemiológia a etiológia
WM je veľmi vzácne ochorenie, tvorí približne 1 – 2 % z celkového počtu hematologických malignít. Štatistické údaje výskytu ochorenia na Slovensku nemáme.
Údaje z USA: Incidencia upravená na vek je 3,4/milión mužov a 1,7/milión žien (20). Údaje z Európy: 7,3/milión mužov a 4,2/milión žien (9).
Vyššia incidencia je v kaukazskej populácii než v afro‑americkej. WM je chorobou starších ľudí. Medián veku v čase diagnózy je 63 – 75 rokov.
Pre vznik WM neexistuje žiadna definitívna etiológia. Boli hlásené environmentálne, familiárne, genetické a vírusové faktory (18). U prvostupňových príbuzných pacientov s WM bolo zistené až 20-násobne zvýšené riziko rozvoja WM (a tiež zvýšené riziko, ale nižšie pre iné B‑lymfoproliferatívne choroby) (9). Z genetických a epigenetických zmien je najvýznamnejšia (až v 55 % prípadov) delécia dlhého ramienka 6. chromozómu. Táto del 6q je spojená s horšou prognózou, s vyššou hladinou beta-2-mikroglobulínu a M‑IgM, s anémiou a hypalbuminémiou. Asymptomatická WM s del 6q častejšie progreduje do symptomatickej formy WM, ktorá vyžaduje liečbu. Boli opísané aj ďalšie cytogenetické zmeny, ale nie sú pre WM špecifické (del 13q, trizómia 4 a 8). Mutácia MYD88 (Myeloid differentiation primary response gene 88) bola zistená až u 90 % pacientov s WM. Mutovaný proteín MYD88L265P zlepšuje prežívanie malígnych buniek. Stanovenie MYD88 sa môže uplatniť v diagnostike a prognóze, ale aj v odlíšení WM od ostatných IgM monoklonových gamapatií (7). Podobne prognostický význam môže mať aj prítomnosť ďalšej mutácie – CXCR4(WHIM) (8).
Najvýznamnejším rizikovým faktorom vzniku WM je monoklonová gamapatia nejasného významu IgM (IgM‑MGUS). Pravdepodobnosť progresie je 1 – 2 % ročne.
Klinický obraz
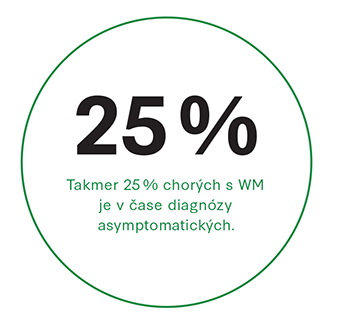
Takmer 25 % chorých s WM je v čase diagnózy asymptomatických. Klinické prejavy vyplývajú:
z infiltrácie kostnej drene patologickými bunkami (anémia, trombocytopénia),
z toxického pôsobenia monoklonového imunoglobulínu IgM (hyperviskozita),
z postihnutia iných orgánov patologickými bunkami (lymfadenopatia, splenomegália).
Na začiatku ochorenia sú nešpecifické, tzv. B‑príznaky:
chronická únava, ktorá neustupuje ani po dostatočnom oddychu, sprievodným javom je pocit slabosti a nevýkonnosti,
nechutenstvo a chudnutie,
febrility či subfebrility.
Patologická únava súvisí s anémiou a hyperviskozitou. Samotná anémia môže mať viac príčin (infiltrácia kostnej drene s útlakom fyziologickej krvotvorby, útlm erytropoézy prozápalovými cytokínmi, hemodilúcia pri vysokej koncentrácii IgM, krvné straty, ale aj chronická AIHA) (3).
Hyperviskozita spôsobuje zvýšené krvácanie zo slizníc nosa, ďasien, tráviaceho traktu. Môže zapríčiniť poruchy zraku: krvácanie do sietnice, ale aj trombózu retinálnych ciev, exsudáty a edém papily. To môže viesť k bolestiam hlavy, závratom, dvojitému videniu. Dôsledkom hyperviskozity môžu byť aj poruchy sluchu. Výnimočne môže vyústiť do kómy, či do krvácania do mozgu. Zvýšená viskozita krvi a zvýšený plazmatický objem môžu spôsobiť zlyhanie srdca.
Pomerne častým nálezom pri WM je kryoglobulinémia. Kryoglobulinémia I. typu je spôsobená monoklonovým IgM, ktorý v chlade gelifikuje a spôsobuje poruchy prekrvenia končatín vystavených chladu. Môže sa prejaviť ako chladová urtika, ale tiež aj ako purpura. Najťažšími prejavmi sú kožné nekrózy, ulcerácie, ale aj poškodenie obličiek. Kryoglobulinémia II. typu je imunokomplexová choroba, kryoglobulín je komplex monoklonového IgM a polyklonového imunoglobulínu typu IgG. Má charakter reumatoidného faktora. Spôsobuje vaskulitídy najmä malých ciev kože, obličiek, pečene a periférnych nervov. Typickým prejavom je purpura (3).
Chladové aglutiníny sú autoprotilátky, ktoré sa viažu na antigény erytrocytov. Po naviazaní chladovej monoklonovej protilátky typu IgM na erytrocyty, dochádza k hemolýze erytrocytov, zhoršuje sa pri vystavení chladu.
Neuropatia spôsobená monoklonovým IgM s protilátkovou aktivitou namierenou proti antigénom nervových vlákien má najčastejšie charakter distálnej senzorickej demyelinizačnej neuropatie. Častá je ataxia, ľahká slabosť, občas aj tras.
Amyloidóza vzniká ukladaním depozitov monoklonového IgM v orgánoch, najmä v srdci, v periférnych nervoch, v obličkách ale aj v mäkkých tkanivách, v pečeni a v pľúcach. U niektorých osôb sa môžu objaviť noduly, papuly a makuly na koži, tvorené depozitmi monoklonového IgM.
Diagnostika
WM je dnes presne definovaná prítomnosťou lymfoplazmocytového lymfómu a monoklonového imunoglobulínu typu M. V definícii choroby nie je uvedená koncentrácia M‑IgM, pretože malígne bunky pravdepodobne produkujú niekedy viac a niekedy menej M‑IgM. Koncentrácia M‑IgM má malý alebo žiadny prognostický význam (3).
Diagnóza WM sa opiera:
o histologický dôkaz (trepanobiopsiou) klonálnej infiltrácie kostnej drene lymfoplazmocytmi, intertrabekulárny typ infiltrácie kostnej drene,
o zistenie M‑IgM v sére (imunoelektroforézou) a jeho kvantitatívne stanovenie,
ak sú indikované: kryoglobulíny, chladové aglutiníny, von Willebrandtov skríning, proteinúria za 24 hodín,
trepanobiopsia a aspirácia kostnej drene
imunohistochémia,
Imunofenotypizácia,
FISH pre mutáciu génu MYD88 L265P.
4. CT trupu (hrudník, brucho, malá panva) s kontrastom
Diferenciálna diagnóza
Treba vylúčiť zriedkavú sekundárnu (symptomatickú) makroglobulinémiu IgM pri systémových procesoch (zápalových, autoimunitných, pri cirhóze) alebo pri karcinómoch (16). U detí a adolescentov sa môže vyskytnúť primárny vrodený hyper‑IgM syndróm z genetických príčin, spojený s častým výskytom oportunistických infekcií (16).
Monoklonový IgM sa môže vyskytovať pri lymfoproliferatívnych ochoreniach, zvyčajne zo skupiny nízko agresívnych chorôb (chronická B‑lymfocytová leukémia, vlasatobunková leukémia, splenický lymfóm) ale aj pri zriedkavom mnohopočetnom IgM myelóme a IgM amyloidóze. Koncentrácia IgM pri týchto ochoreniach obvykle nepresahuje 30 g/l M‑IgM. Nad 30 g/l bola pozorovaná výlučne u pacientov s WM, ale väčšina pacientov s WM mala koncentráciu M‑IgM pod 30 g/l (3).
Veľmi dôležité je odlíšiť monoklonovú gamapatiu nejasného významu typu IgM (IgM‑MGUS). Je prítomný M‑IgM zvyčajne do 30 g/l, v kostnej dreni nie sú prítomné malígne bunky, nie je prítomná lymfadenopatia a nie sú prítomné príznaky poškodenia organizmu monoklonovým IgM.
Ak je prítomný monoklonový IgM a súčasne je diagnostikovaná kryoglobulinémia, choroba chladových aglutinínov, neuropatia alebo amyloidóza a v kostnej dreni nie je prítomná malígna lymfoproliferácia, radia sa tieto prípady do skupiny nazvanej choroby spôsobené monoklonovým imunoglobulínom – monoclonal IgM related disorders (6). M‑IgM je v týchto prípadoch produkovaný malým množstvom klonálnych buniek, ktoré nie sú ešte detekovateľné morfologicky (3).
Ak je histologicky potvrdená infiltrácia kostnej drene lymfoplazmocytovým lymfómom a monoklonový IgM a zároveň nie sú prítomné žiadne známky choroby, hovoríme o asymptomatickej forme WM (Tabuľka č. 2) (3).
Prognóza
WM je choroba s pomerne priaznivou prognózou. Podľa údajov z Európskeho registra v rokoch 2000 – 2014 bol medián celkového prežitia viac než 10 rokov (10). Osud chorých je ale veľmi rôznorodý. Informácie o prognóze poskytujú prognostické indexy. Najčastejšie používaný je prognostický skórovací systém uvedený v Tab. č. 3. Zvýšená hladina laktátdehydrogenázy v kombinácii s vysokým rizikom IPSSWM definuje pacientov s obzvlášť nepriaznivou prognózou (10).
Liečba
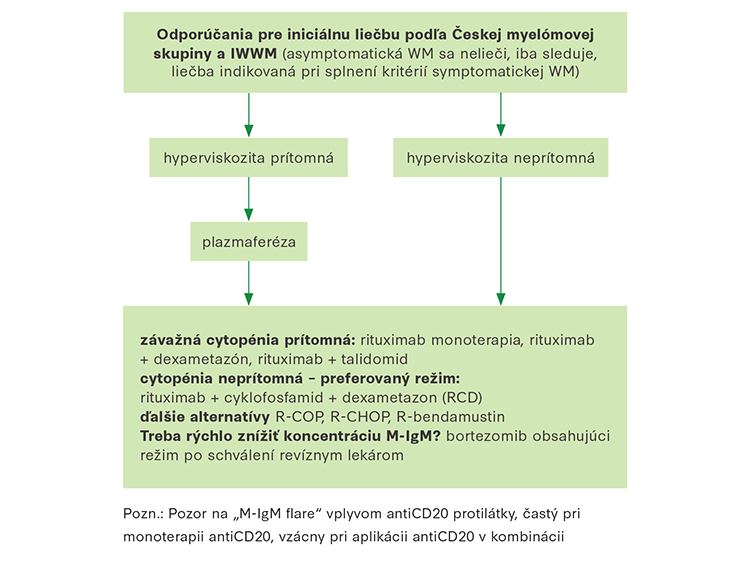
Asymptomatická forma WM sa nelieči. Jedným z dôvodov je identická úmrtnosť u asymptomatických chorých ako u bežnej populácie (10). Prechod do symptomatickej formy WM je kontinuálny. Pravdepodobnosť progresie z asymptomatickej do symptomatickej formy WM po 1, 3, 5 a 10 rokoch bola 6 %, 39 %, 59 % a 68 % (1). Zvýšené riziko transformácie signalizujú vyššie koncentrácie beta-2-mikroglobulínu, nižšia koncentrácia hemoglobínu, výška koncentrácie M‑IgM a miera infiltrácie kostnej drene (1). Indikácie na začatie liečby uvádza Tabuľka č. 4.
Plazmaferéza
Pri koncentrácii M‑IgM nad 40 g/l sa odporúča vždy vyšetriť očné pozadie. Ak by sa našli zmeny na očnom pozadí poukazujúce na hyperviskozitu alebo pri iných príznakoch hyperviskozity je vhodné urobiť veľkoobjemovú liečebnú plazmaferézu s náhradou odobratej plazmy albumínom a mrazenou plazmou (1).
Paliatívna perorálna liečba
Ak pacient nie je schopný podstúpiť intenzívnejšiu liečbu, je možné podať chlorambucil alebo fludarabin v tabletkách.
Monoklonové protilátky
Ak je pacient schopný podstúpiť komplexnú liečbu, štandardnou súčasťou liečby je dnes anti‑CD20 protilátka, rituximab, pretože nemá dlhodobú toxicitu a jej použitie neblokuje neskorší zber kmeňových buniek krvotvorby z periférnej krvi. V monoterapii dosahuje síce len 30-percentnú parciálnu liečebnú odozvu (50 % redukcie M‑IgM). V kombinácii s ďalšími liekmi s dokázanou účinnosťou pri WM sa zásadne zvyšuje efekt liečby. Za optimálnu liečbu 1. línie sa v posledných rokoch považuje kombinácia rituximabu s cyklofosfamidom a dexametazónom (RCD). Touto kombináciou dochádza až k 83-percentnej liečebnej odozve. 2-ročné bezpríznakové obdobie bolo dosiahnuté v 67 % prípadov. V publikácii IMWW túto kombináciu považujú za optimálnu pre liečbu 1. línie (1). Klasická R‑CHOP (rituximab, cyklofosfamid, doxorubicín, vincristin a prednison) predstavuje alternatívu k liečbe RCD v 1. línii. Dosahuje najmenej 90-percentnú mieru liečebnej odozvy, ale za cenu vyššej toxicity než režim RCD.
Purínové analógy
Fludarabin je veľmi účinným liekom pri WM. Monoterapia fludarabinom viedla k dlhšiemu bezpríznakovému i celkovému prežívaniu v porovnaní s chlorambucilom. Kombinácia rituximabu a fludarabinu dosahuje vysokú mieru liečebnej odozvy, až 95 %, s mediánom trvania liečebnej odpovede 51,2 mesiacov. Liečebné postupy obsahujúce kombináciu fludarabinu, alkylačného cytostatika a rituximabu viedli síce k rýchlemu nástupu liečebnej odpovede, ale sú spojené s rizikom vzniku myelodysplastického syndrómu a transformácie do agresívneho typu lymfómu. Od roku 2015 už nie sú štandardne odporúčané pre liečbu 1. línie, ale ich použitie sa zvažuje pri skorom relapse choroby (4).
Nové lieky pre WM
Bendamustin je veľmi účinným liekom pre WM. Mohol by nahradiť cyklofosfamid v kombinácii s dexametazónom a rituximabom, ak by bol pre týchto pacientov dostupný (10). Bendamustin je vhodným liekom na iniciálnu liečbu, aj na liečbu relapsu.
Bortezomib, inhibítor proteazómu, je vysoko účinný liek aj pre WM. Liečebná odpoveď po bortezomibe nastupuje rýchlejšie, než po iných režimoch. Medzinárodné odporúčania IWWM z roku 2014 uprednostňujú podávať bortezomib 1-krát týždenne namiesto 2-krát a uprednostňujú podkožné podanie pred intravenóznym s cieľom znížiť riziko vzniku závažnej neuropatie. Autori IWWM odporúčajú použiť liečebné protokoly obsahujúce bortezomib už v rámci prvej línie liečby v prípade hyperviskozity a v prípadoch, keď treba veľmi rýchlo navodiť liečebnú odpoveď, a teda odstrániť symptómy choroby.
Carfilzomib je novší inhibítor proteazómu, jeho podanie nespôsobuje neuropatiu.
Talidomid a lenalidomid. Rituximab v kombinácii s talidomidom dosiahol 72-percentnú liečebnú odpoveď, rituximab v kombinácii s lenalidomidom 50 %. Nevýhodou lenalidomidu je, že prehlbuje v priebehu liečby anémiu a talidomid často spôsobuje neuropatiu.
Ibrutinib, inhibítor Brutonovej kinázy. Prvá skúsenosť s ibrutinibom v súbore chorých už predtým liečených, miera liečebnej odpovede dosiahla 90,5 %, aspoň parciálnu remisiu dosiahlo 73 % pacientov. Miera liečebnej odozvy bola výrazne ovplyvnená mutačným stavom. U pacientov, ktorí boli MYD88(L265P), CXCR4(WT), bola dosiahnutá 100-percentná liečebná odozva a z toho v 91,2 % aspoň parciálna liečebná odpoveď. 2-ročné bezpríznakové prežitie a 2-ročné celkové prežívanie dosiahlo 69,1 – 95,2 % (10). Liečba je pomerne dobre tolerovaná. Z nežiaducich účinkov bol zaznamenaný vznik fibrilácie predsiení.
Vysokodávkovaná chemoterapia s autológnou transplantáciou krvotvorných buniek je účinnou liečbou pre vhodných pacientov, v súčasnosti je indikovaná na liečbu prvého relapsu.
Záver
Napriek tomu, že ochorenie je veľmi vzácne, v posledných rokoch sa urobili významné pokroky v odhalení patofyziológie ochorenia, najmä v objavení mutácií génov MYD88 a CXCR4 u pacientov s WM. Dajú sa využiť v diferenciálnej diagnostike, v stanovení prognózy a liečebných postupov. Prehĺbili sa vedomosti o autoimunitných prejavoch M‑IgM, ktorých správne rozpoznanie môže viesť k rozhodnutiu o začatí liečby. Boli tiež publikované štatistické analýzy veľkých súborov pacientov, ktoré poukazujú na zlepšovanie prognózy, výrazne lepšiu prognózu u mladších osôb a prekvapivé zistenie, že vo všetkých vekových kategóriách je vyšší počet úmrtí z iných príčin, než na samotné ochorenie WM (1). Vzhľadom na túto priaznivú prognózu sa v poslednom čase pri výbere liečby dostáva do popredia miera záťaže chorého liečbou. Preto sa zmenili názory a uprednostňuje sa menej agresívna iniciálna liečba. Bol tiež upresnený význam nových liekov používaných pri liečbe WM (bortezomib, bendamustin, imidy, carfilzomib, ibrutinib, ofatumumab) a prebiehajú štúdie s novými liečebnými postupmi – s inhibítormi proteazómu (ixazomib), bcl-2 inhibítormi (venetoclax), druhogeneračnými inhibítormi Brutonovej kinázy (acalabrutinib, zanubrutinib) a cielená protilátková terapia (ulocuplumab) (22).
Literatúra
Adam Z., Pour L., Krejčí M., Ševčíková S., Pourová E., Ševčíková E., Král Z., Mayer J. Změny v prognóze a v léčbě Waldenströmovy makroglobulinemie: přehled literatury a vlastní zkušenosti. Vnitř Lék 2016; 62(1): 25-39.
Adam Z. Waldenströmova makroglobulinemie. In Adam Z. a kol.: Diagnostické a léčebné postupy u maligních chorob, Grada Publishing, Praha, 2004, s. 531-534.
Adam Z. Waldenströmova makroglobulinemie. In Adam Z., Krejčí M., Vorlíček J. a kol.: Hematologie Přehled maligních hematologických nemocí, Grada Publishing, Praha, 2008, s. 263-287.
Adam Z., Hájek R., Krejčí M., Maisnar V. a kol. Diagnostika a léčba Waldenströmovy makroglobulinemie. Transfuze hematol. dnes Supplementum, 2014: 7-22.
Gertz M. A. Waldenström macroglobulinemia treatment algorithm 2018. Blood Cancer Journal 2018; 8:40, dostupné z DOI https://doi.org/10.1038/s41408-018-0076-5.
Grunenberg A., Buske CH. Monoclonal IgM Gammopathy and Waldenström’s Macroglobulinemia. Dtsch Arztebl Int 2017; 114: 745-51.
Haber J. Waldenströmova makroglobulinemie. Lymfómové fórum 2017. Zborník prednášok: 12-21.
Kapoor P., Ansell S. et al. Diagnosis and Managment of Waldenström Macroglobulinemia: Mayo Stratification of Macroglobulinemia and Risk‑Adapted Therapy (mSMART) Guidelines 2016. JAMA Oncol. 2017; 3(9): 1257-1265. doi:10.1001/jamaoncol.2016.5763.
Kastritis E., Leblond V., Dimopoulos M.A. et al. Waldenström macroglobulinaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‑up. Annals of Oncology 2018; 29 (Supplement 4): 41-50. Dostupné z DOI https://doi.org/10.1093/annonc/mdy179.
Kaščák M., Kufová Z., Growková K., Hájek R. Waldenströmova makroglobulinemie v roce 2016: optimalizace imunochemoterapie s cílem minimalizace nežádoucích účinků. Onkológia (bratisl.) 2016; 11(5): 291-295.
Kumar S.K., Callander N. S., Atanackovic D., et al. National Comprehensive Cancer Network. NCCN Guidelines Version 1.2017 Waldenström’s Macroglobulinemia/Lymphoplasmactic Lymphoma Available from: accessed May 15, 2017.
Kutálková K., Sedlaříková L., Adam Z., Ševčíková S. Genetické změny u Waldenströmovy makroglobulinemie. Vnitř Lék. 2016; 62(1): 40-43.
Kyle R.A., Anderson C.K. A Tribute to Jan Gosta Waldenström. Blood 1997; 89: 4245-4247.
Morel P., Duhamel A., Gobbi P. et al. International prognostic scoring system for Waldenström macroglobulinemia. Blood 2009; 113: 4163-4170.
Paludo J., Ansell S. Waldenström macroglobulinemia: biology, genetic, and therapy. Blood and Lymphatic Cancer 2016; 6: 49-58, dostupné z DOI https://doi.org/10.2147/BLCTT.S84157.
Sakalová A. Nádorové ochorenia plazmatických buniek a paraproteinémie. In Sakalová A., Bátorová A., Mistrík M., Hrubiško M. a kol.: Klinická hematológia, Osveta, Martin, 2010, s. 136-138.
Sakalová A. a kol. Súčasná klasifikácia, diagnostika a prognóza primárnych monoklonových gamapatií (paraproteinémií). Transfuze Hematol. dnes, 2010; 16 (4): 193-196.
Seiter K. Waldenström Macroglobulinemia. Dostupné z https://emedicine.medscape.com/article/207097.
Treon SP. How I treat Waldenström macroglobulinemia. Blood 2015; dostupné z DOI https://doi.org/10.1182/blood-2015-01-553974.
Wang H., CHen Y, Li F et al. Temporal and geografic variations of Waldenström macroglobulinemia incidence. A large population based study. Cancer 2012; 118: 3793-3800.
Yun S., Johnson A. et al. Waldenström Macroglobulinemia: Review of pathogenesis and Managment. Clin Lymphoma Myeloma Leuk. 2017; 17 (5): 252-262. doi:10.1016/j.clml.2017. 02. 028.
Harrison P. New Drugs Poised to Expand Treatment Horizons in WM. Medscape 2018, https://www.medscape.com/viewarticle/897258.
Tento článok sa nachádza v čísle invitro 01/2019
Lymfatické uzliny
Jarné vydanie časopisu inVitro prináša aktuálne informácie o lymfatickom systéme. Okrem odborných textov zaoberajúcich sa jeho ochoreniami a ich liečbou v ňom už tradične nájdete…